Chapter 2 Exploration Diagnostic
2.1 Setup
First, we need to do some set up to analyze our data
Include dependencies
library(ggplot2) # For plotting
library(tidyverse) # For data wrangling
library(knitr) # For making nice rmarkdown documents
library(cowplot) # For theme
library(viridis) # For color scale
library(RColorBrewer) # For more color scales
library(rstatix)
library(ggsignif) # For adding pairwise significance to plots
library(Hmisc) # For bootstrapping condifence internvals
library(kableExtra) # For displaying nice tables
source("https://gist.githubusercontent.com/benmarwick/2a1bb0133ff568cbe28d/raw/fb53bd97121f7f9ce947837ef1a4c65a73bffb3f/geom_flat_violin.R") # For raincloud plots
library(readr) # For reading in data
library(stringr) # For manipulating string data
library(ggpubr) # For displaying correlation statistics on plots
library(infotheo) # For causality analysis
library(scales) # For displaying scales nicely in facetted plots
library(osfr) # For downloading the data for this projectThese analyses were conducted in the following computing environment:
## _
## platform x86_64-apple-darwin17.0
## arch x86_64
## os darwin17.0
## system x86_64, darwin17.0
## status
## major 4
## minor 1.1
## year 2021
## month 08
## day 10
## svn rev 80725
## language R
## version.string R version 4.1.1 (2021-08-10)
## nickname Kick ThingsSetup constants to be used across plots
# Labeler for stats annotations
p_label <- function(p_value) {
threshold = 0.0001
if (p_value < threshold) {
return(paste0("p < ", threshold))
} else {
return(paste0("p = ", p_value))
}
}
# Significance threshold
alpha <- 0.05
# Common graph variables
performance_ylim <- 1
coverage_ylim <- 1.0
####### misc #######
# Configure our default graphing theme
theme_set(theme_cowplot())The data for this project are hosted on osf.io. The following chunk downloads them automatically if they haven’t already been downloaded.
# Read in data
osf_retrieve_file("esm4r") %>% osf_download(conflicts = "skip") # Download data from osf
data_loc <- "final_exploration_diagnostic_data.csv"
data <- read_csv(data_loc, na=c("NONE", "NA", ""))
## Clean up data columns
# Make selection name column human readable
data <- data %>% mutate(selection_name = as.factor(case_when(
selection_name == "EpsilonLexicase" ~ "Lexicase",
TOUR_SIZE == 1 ~ "Random",
selection_name == "Tournament" ~ "Tournament",
selection_name == "FitnessSharing" ~ "Fitness Sharing",
selection_name == "EcoEa" ~ "EcoEA"
)))
# Calculate performance statistics.
# Elite trait avg is the avg per-site performance of the best individual
data$elite_trait_avg <-
data$ele_agg_per / data$OBJECTIVE_CNT
data$unique_start_positions_coverage <-
data$uni_str_pos / data$OBJECTIVE_CNT
# Convert elite_trait_avg to percent of maximum possible
data$elite_trait_avg <- data$elite_trait_avg/data$TARGET
# Grab data from just the final time point
final_data <- filter(data, evaluations==max(data$evaluations))2.2 Performance
For context, it’s important to know how each selection scheme performed on the exploration diagnostic.
2.2.1 Over time
First we look at the dynamics of performance over time.
2.2.1.1 Trait performance
Here, we plot average trait performance (i.e. fitness) over time for each selection scheme. We log the x-axis because Eco-EA gains fitness over a very long time scale, whereas the interesting dynamics for the other selection schemes occur relatively quickly.
ggplot(
data,
aes(
x=gen, # Generations
y=elite_trait_avg, # Performance
color=selection_name, # Selection scheme
fill=selection_name
)
) +
stat_summary(geom="line", fun=mean) + # Plot line showing mean for each selection scheme
stat_summary( # Add shading around each line indicating 95% confiedence interval
geom="ribbon",
fun.data="mean_cl_boot",
fun.args=list(conf.int=0.95),
alpha=0.2,
linetype=0
) +
scale_y_continuous(
name="Average trait performance", # Set y axis title
limits=c(0, performance_ylim) # Set y axis range to include all possible performance values
) +
scale_x_log10( # Log x axis
name="Generation" # Set x axis title
) +
scale_color_discrete("Selection") + # Set legend title
scale_fill_discrete("Selection") # Set legend title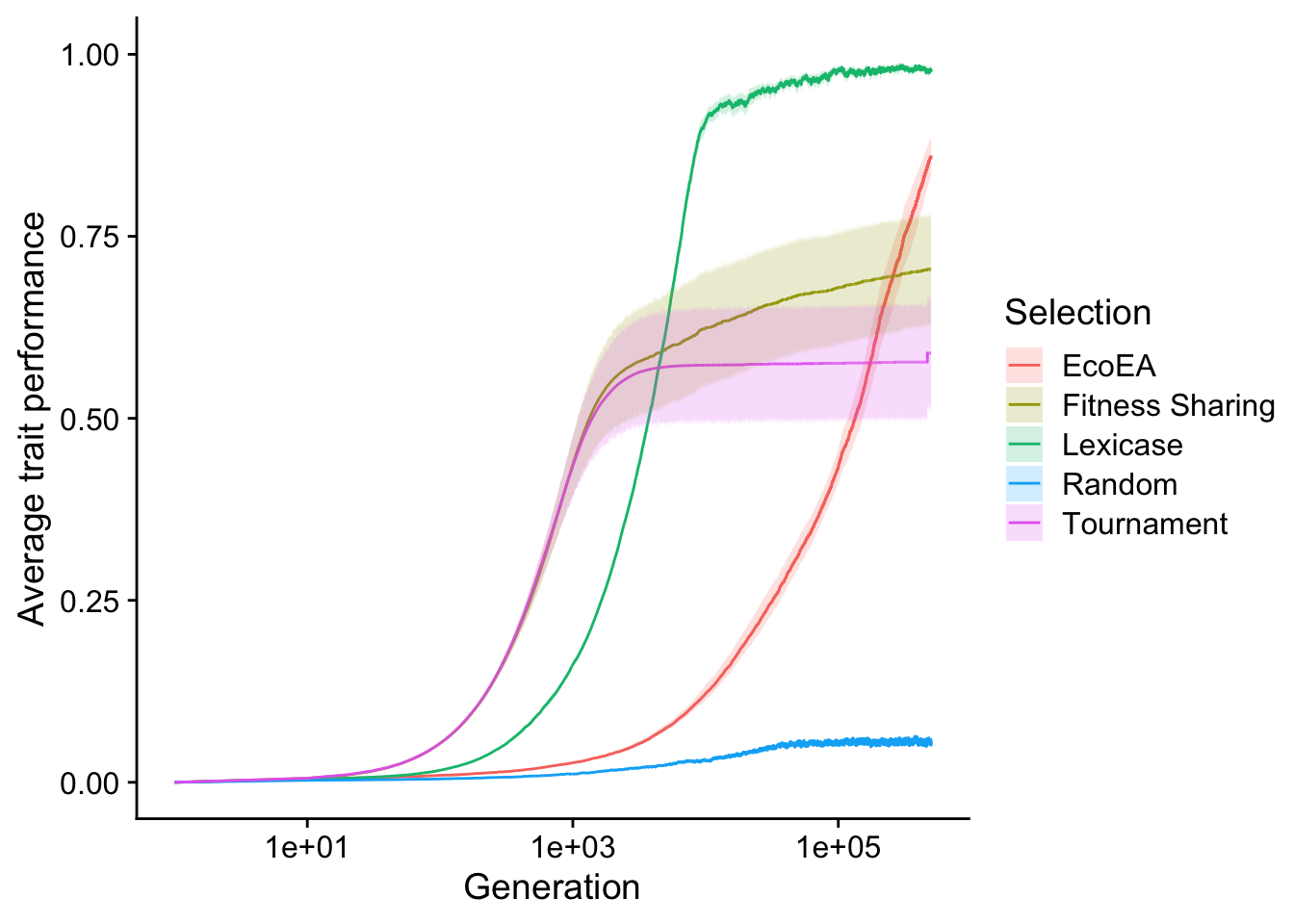
As observed by Hernandez et al. in their original paper on the exploration diagnostic (Hernandez, Lalejini, and Ofria 2021), fitness in tournament selection initially increases quickly and then plateaus. Fitness in lexicase selection increases slightly slower but plateaus at a much higher value (nearly 100%). Fitness sharing behaves similarly to tournament selection, but maintains a slight upward trajectory (note that, because the x axis is on a log scale, this trajectory is very gradual). Eco-EA takes substantially longer to increase in fitness but ultimately surpasses fitness sharing and tournament selection. It is unclear whether it would pass lexicase selection if these runs were allowed to continue for slightly longer; they do not appear to have plateaued yet. We chose to cut them off at 500,000 generations due to resource constraints and the fact that the questions we’re asking here are not really about final fitness.
2.2.1.2 Activation position coverage
Out of curiosity, we also ran the analysis of unique activation positions present in the population, used by Hernandez et. al. This analysis tells us about the diversity of start positions for the coding region represented in the population. As the set of start positions in the population tends to represent a meaningful constraint on the number of paths through the fitness landscape that are currently accessible, this is in some sense a metric of useful diversity in the population
ggplot(data, aes(x=gen, y=unique_start_positions_coverage, color=selection_name, fill=selection_name)) +
stat_summary(geom="line", fun=mean) +
stat_summary(
geom="ribbon",
fun.data="mean_cl_boot",
fun.args=list(conf.int=0.95),
alpha=0.2,
linetype=0
) +
scale_y_continuous(
name="Activation position coverage"
) +
scale_x_log10(
name="Generation"
) +
scale_color_discrete("Selection")+
scale_fill_discrete("Selection")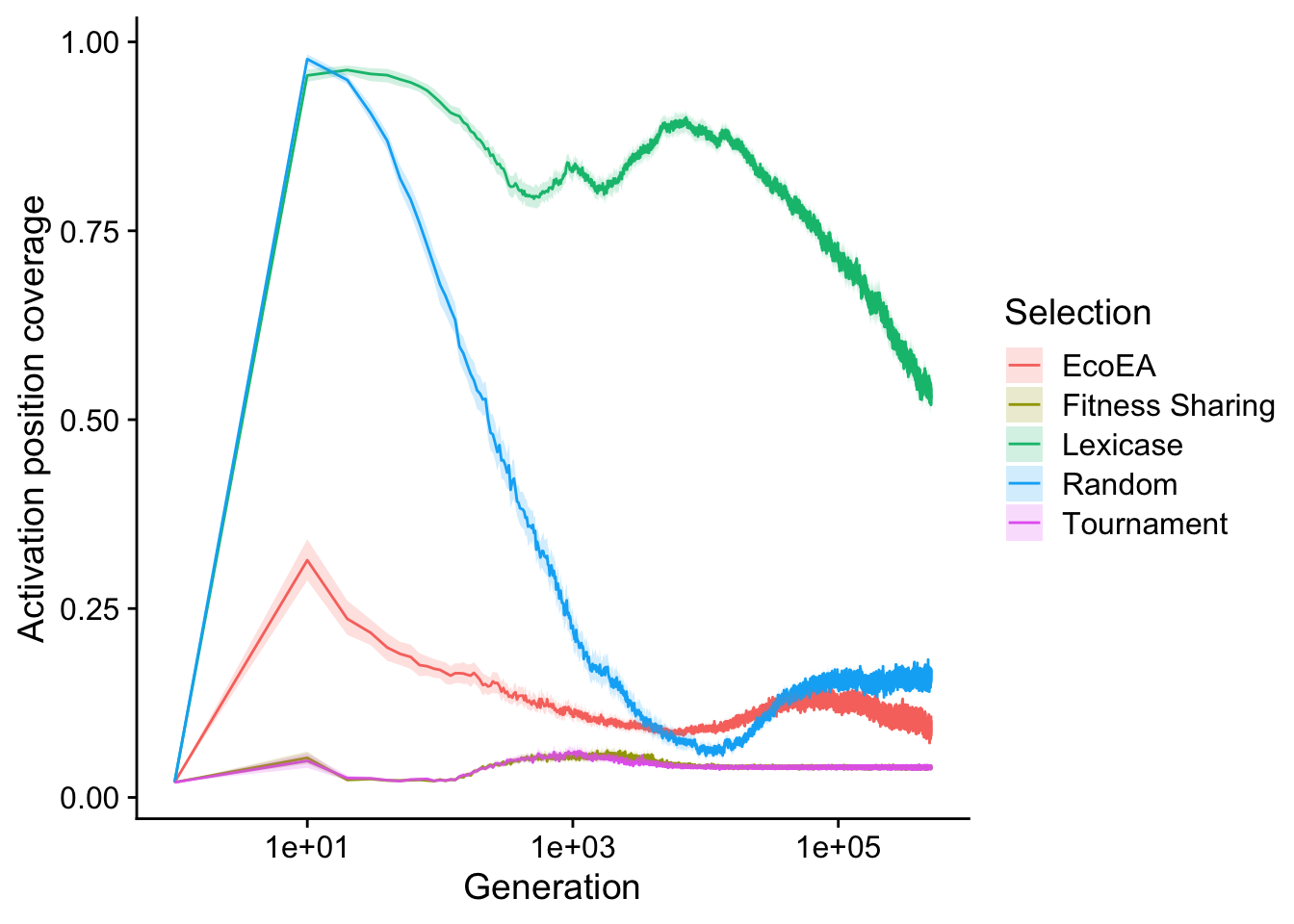
We see that lexicase selection maintains by far that largest number of unique start positions, even surpassing the number maintained by random drift. This suggests that lexicase selection is actively selecting for maintaining a diversity of start positions. Tournament selection and fitness sharing perform virtually identically, with Eco-EA falling in between.
2.2.2 Final
While trends over time are more informative, it can be hard to visualize the full distribution (particularly the extent of variation). Thus, we also conduct a more detailed analysis of performance at the final time point.
2.2.2.1 Trait performance
First we conduct statistics to identify which selection schemes are significantly different from each other.
# Compute manual labels for geom_signif
stat.test <- final_data %>%
wilcox_test(elite_trait_avg ~ selection_name) %>%
adjust_pvalue(method = "bonferroni") %>% # Apply Bonferroni correction for multiple comparisons
add_significance() %>%
add_xy_position(x="selection_name",step.increase=1)
stat.test$label <- mapply(p_label,stat.test$p.adj)Then we make raincloud plots (Allen et al. 2021) of each selection scheme.
elite_final_performance_fig <- ggplot(
final_data,
aes(
x=selection_name,
y=elite_trait_avg,
fill=selection_name
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
scale="width"
) +
geom_point(
mapping=aes(color=selection_name),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
name="Average trait performance",
limits=c(0, performance_ylim)
) +
scale_x_discrete(
name="Selection"
) +
scale_fill_discrete(
name="Selection"
) +
scale_color_discrete(
name="Selection"
) +
theme(legend.position="none")
elite_final_performance_fig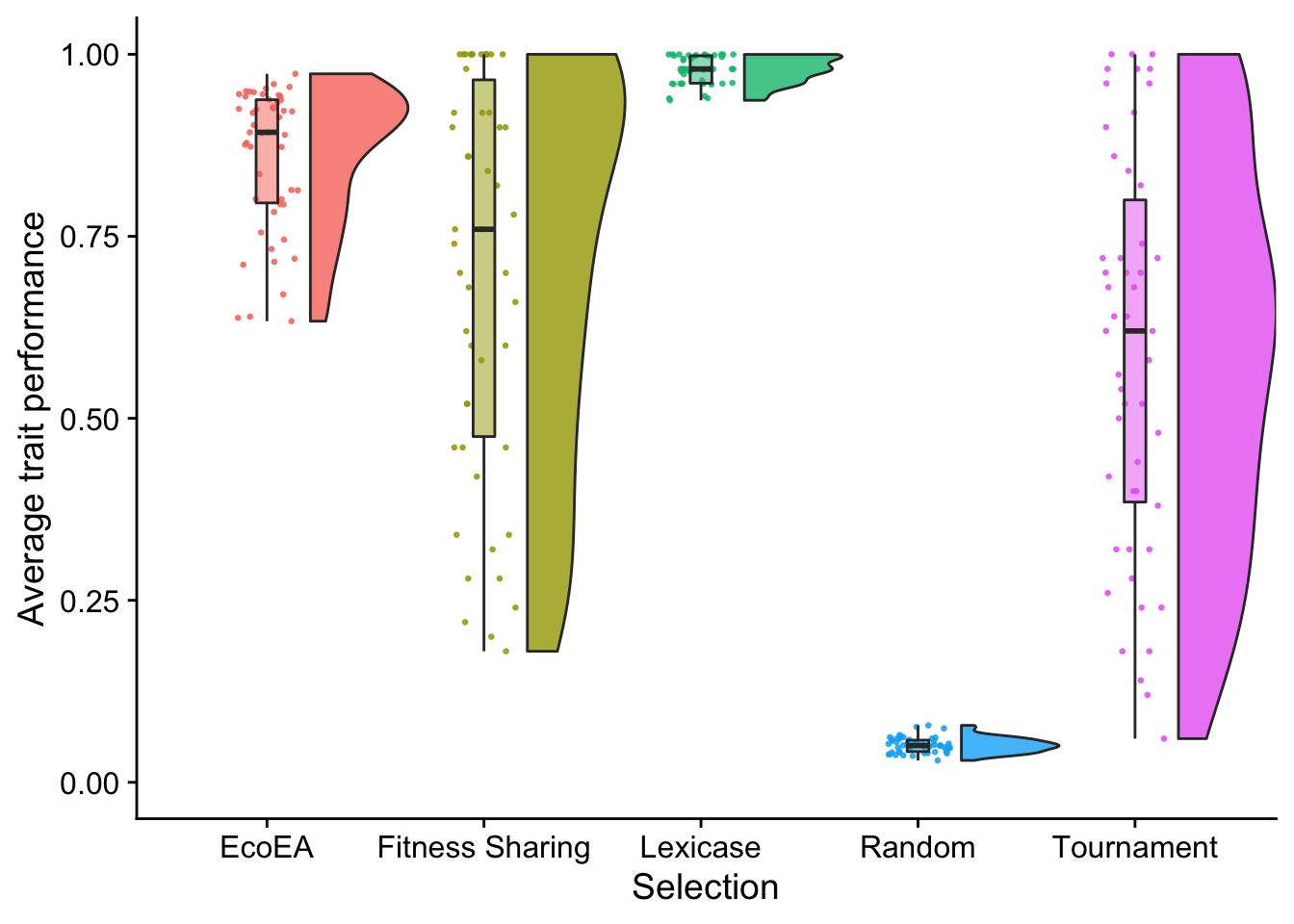
These observations look fairly consistent with the timeseries plots.
Next, we output the results of our significance testing.
stat.test %>%
kbl() %>%
kable_styling(
bootstrap_options = c(
"striped",
"hover",
"condensed",
"responsive"
)
) %>%
scroll_box(width="600px")| .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | y.position | groups | xmin | xmax | label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| elite_trait_avg | EcoEA | Fitness Sharing | 50 | 50 | 1561 | 3.20e-02 | 3.20e-01 | ns | 1.922000 | EcoEA , Fitness Sharing | 1 | 2 | p = 0.32 |
| elite_trait_avg | EcoEA | Lexicase | 50 | 47 | 60 | 0.00e+00 | 0.00e+00 | **** | 2.946444 | EcoEA , Lexicase | 1 | 3 | p < 1e-04 |
| elite_trait_avg | EcoEA | Random | 50 | 50 | 2500 | 0.00e+00 | 0.00e+00 | **** | 3.970889 | EcoEA , Random | 1 | 4 | p < 1e-04 |
| elite_trait_avg | EcoEA | Tournament | 50 | 50 | 1939 | 2.10e-06 | 2.07e-05 | **** | 4.995333 | EcoEA , Tournament | 1 | 5 | p < 1e-04 |
| elite_trait_avg | Fitness Sharing | Lexicase | 50 | 47 | 593 | 2.69e-05 | 2.69e-04 | *** | 6.019778 | Fitness Sharing, Lexicase | 2 | 3 | p = 0.000269 |
| elite_trait_avg | Fitness Sharing | Random | 50 | 50 | 2500 | 0.00e+00 | 0.00e+00 | **** | 7.044222 | Fitness Sharing, Random | 2 | 4 | p < 1e-04 |
| elite_trait_avg | Fitness Sharing | Tournament | 50 | 50 | 1549 | 4.00e-02 | 4.00e-01 | ns | 8.068667 | Fitness Sharing, Tournament | 2 | 5 | p = 0.4 |
| elite_trait_avg | Lexicase | Random | 47 | 50 | 2350 | 0.00e+00 | 0.00e+00 | **** | 9.093111 | Lexicase, Random | 3 | 4 | p < 1e-04 |
| elite_trait_avg | Lexicase | Tournament | 47 | 50 | 2098 | 0.00e+00 | 0.00e+00 | **** | 10.117556 | Lexicase , Tournament | 3 | 5 | p < 1e-04 |
| elite_trait_avg | Random | Tournament | 50 | 50 | 10 | 0.00e+00 | 0.00e+00 | **** | 11.142000 | Random , Tournament | 4 | 5 | p < 1e-04 |
Fitness sharing did not perform significantly differently from Eco-EA or Tournament selection, but all other selection schemes are significantly different.
2.2.2.2 Final activation position Coverage
Now we do the same analysis for final activation position coverage.
First we calculate the statistics
# Compute manual labels for geom_signif
stat.test <- final_data %>%
wilcox_test(unique_start_positions_coverage ~ selection_name) %>%
adjust_pvalue(method = "bonferroni") %>%
add_significance() %>%
add_xy_position(x="selection_name",step.increase=1)
stat.test$manual_position <- stat.test$y.position * 1.05
stat.test$label <- mapply(p_label,stat.test$p.adj)Then we make raincloud plots
unique_start_positions_coverage_final_fig <- ggplot(
final_data,
aes(
x=selection_name,
y=unique_start_positions_coverage,
fill=selection_name
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
scale="width"
) +
geom_point(
mapping=aes(color=selection_name),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
name="Activation position coverage",
limits=c(0, coverage_ylim)
) +
scale_x_discrete(
name="Selection"
) +
scale_fill_discrete(
name="Selection"
) +
scale_color_discrete(
name="Selection"
) +
theme(
legend.position="none"
)
unique_start_positions_coverage_final_fig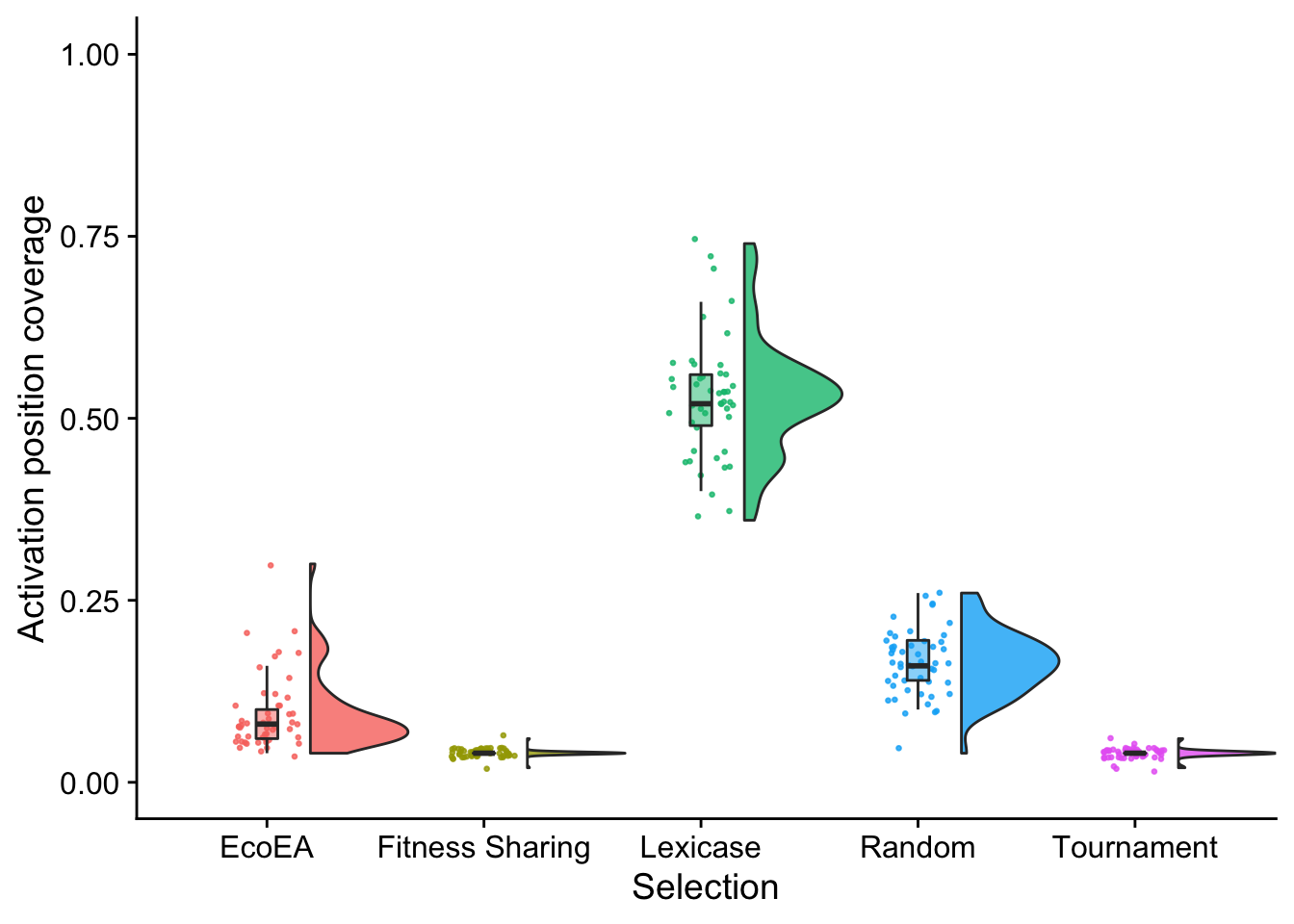
These also look unsurprising.
Lastly, we output the results of significance testing.
stat.test %>%
kbl() %>%
kable_styling(
bootstrap_options = c(
"striped",
"hover",
"condensed",
"responsive"
)
) %>%
scroll_box(width="600px")| .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | y.position | groups | xmin | xmax | manual_position | label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| unique_start_positions_coverage | EcoEA | Fitness Sharing | 50 | 50 | 2392.5 | 0.000 | 0 | **** | 1.420000 | EcoEA , Fitness Sharing | 1 | 2 | 1.491000 | p < 1e-04 |
| unique_start_positions_coverage | EcoEA | Lexicase | 50 | 47 | 0.0 | 0.000 | 0 | **** | 2.175556 | EcoEA , Lexicase | 1 | 3 | 2.284333 | p < 1e-04 |
| unique_start_positions_coverage | EcoEA | Random | 50 | 50 | 339.0 | 0.000 | 0 | **** | 2.931111 | EcoEA , Random | 1 | 4 | 3.077667 | p < 1e-04 |
| unique_start_positions_coverage | EcoEA | Tournament | 50 | 50 | 2387.0 | 0.000 | 0 | **** | 3.686667 | EcoEA , Tournament | 1 | 5 | 3.871000 | p < 1e-04 |
| unique_start_positions_coverage | Fitness Sharing | Lexicase | 50 | 47 | 0.0 | 0.000 | 0 | **** | 4.442222 | Fitness Sharing, Lexicase | 2 | 3 | 4.664333 | p < 1e-04 |
| unique_start_positions_coverage | Fitness Sharing | Random | 50 | 50 | 25.0 | 0.000 | 0 | **** | 5.197778 | Fitness Sharing, Random | 2 | 4 | 5.457667 | p < 1e-04 |
| unique_start_positions_coverage | Fitness Sharing | Tournament | 50 | 50 | 1274.5 | 0.708 | 1 | ns | 5.953333 | Fitness Sharing, Tournament | 2 | 5 | 6.251000 | p = 1 |
| unique_start_positions_coverage | Lexicase | Random | 47 | 50 | 2350.0 | 0.000 | 0 | **** | 6.708889 | Lexicase, Random | 3 | 4 | 7.044333 | p < 1e-04 |
| unique_start_positions_coverage | Lexicase | Tournament | 47 | 50 | 2350.0 | 0.000 | 0 | **** | 7.464444 | Lexicase , Tournament | 3 | 5 | 7.837667 | p < 1e-04 |
| unique_start_positions_coverage | Random | Tournament | 50 | 50 | 2475.5 | 0.000 | 0 | **** | 8.220000 | Random , Tournament | 4 | 5 | 8.631000 | p < 1e-04 |
2.3 Phylogenetic diversity
Next, we analyze the behavior of phylogenetic diversity on the exploration diagnostic.
2.3.1 Relationship between different types of pylogenetic diversity
First, to get a big-picture overview, we make correlation matrices of all the different phylogenetic diversity metrics:
final_data %>%
transmute(MinPD=min_phenotype_pairwise_distance,
MeanPD=mean_phenotype_pairwise_distance,
MaxPD=max_phenotype_pairwise_distance,
VarPD=variance_phenotype_pairwise_distance,
MinED = min_phenotype_evolutionary_distinctiveness,
MeanED= mean_phenotype_evolutionary_distinctiveness,
MaxED=max_phenotype_evolutionary_distinctiveness,
VarED=variance_phenotype_evolutionary_distinctiveness,
PD=phenotype_current_phylogenetic_diversity, # See Faith 1992
MRCA=phen_mrca_depth, # Phylogenetic depth of most recent common ancestor
N=phen_num_taxa # Number of taxonomically-distinct phenotypes
) %>%
cor_mat() %>%
pull_lower_triangle() %>%
cor_plot()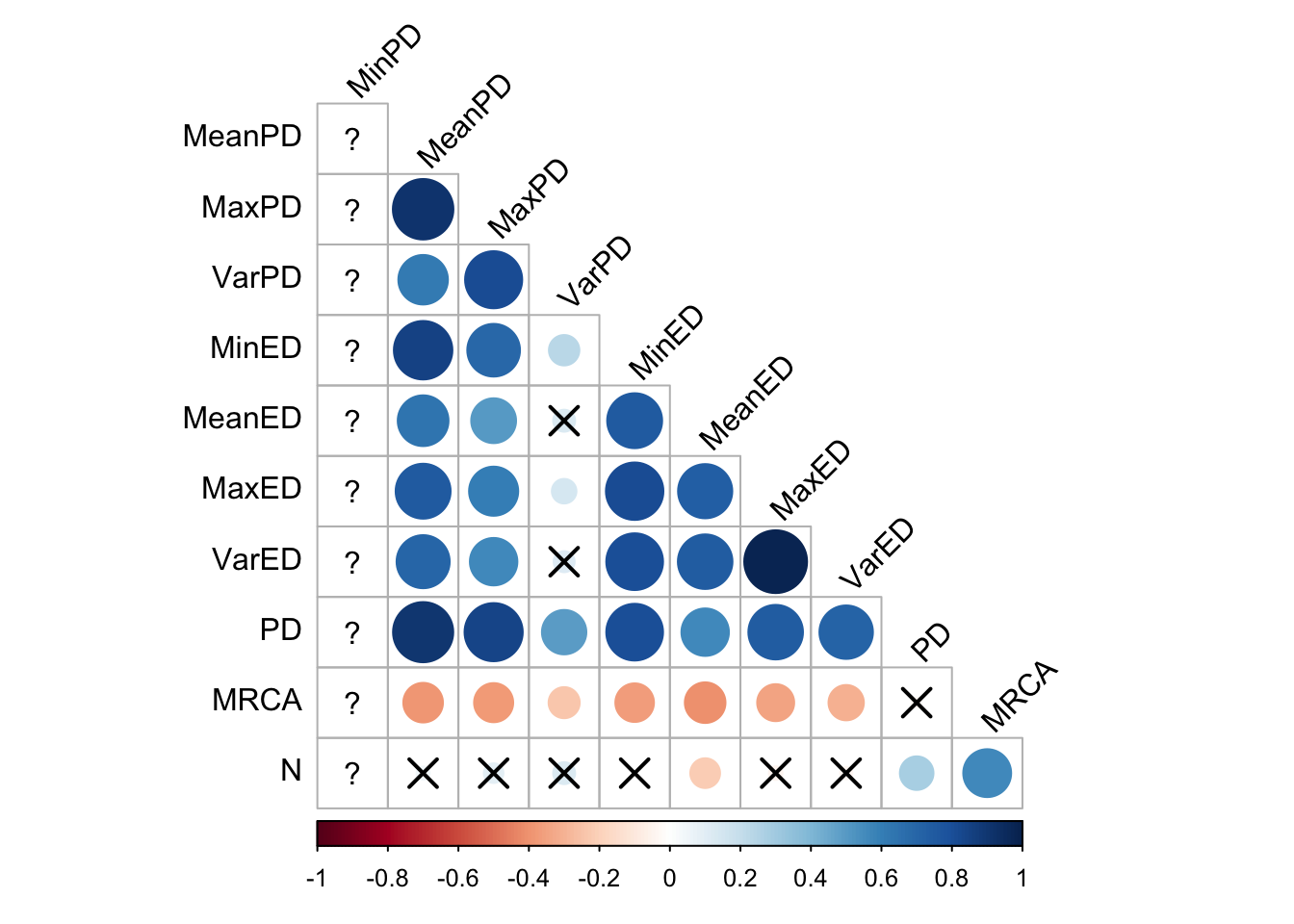
However, these correlations may well vary by selection scheme, and even over time within a selection scheme. Let’s take a look at some scatter plots.
ggplot(
data %>% filter(gen==500000),
aes(
y=mean_phenotype_pairwise_distance,
x=variance_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")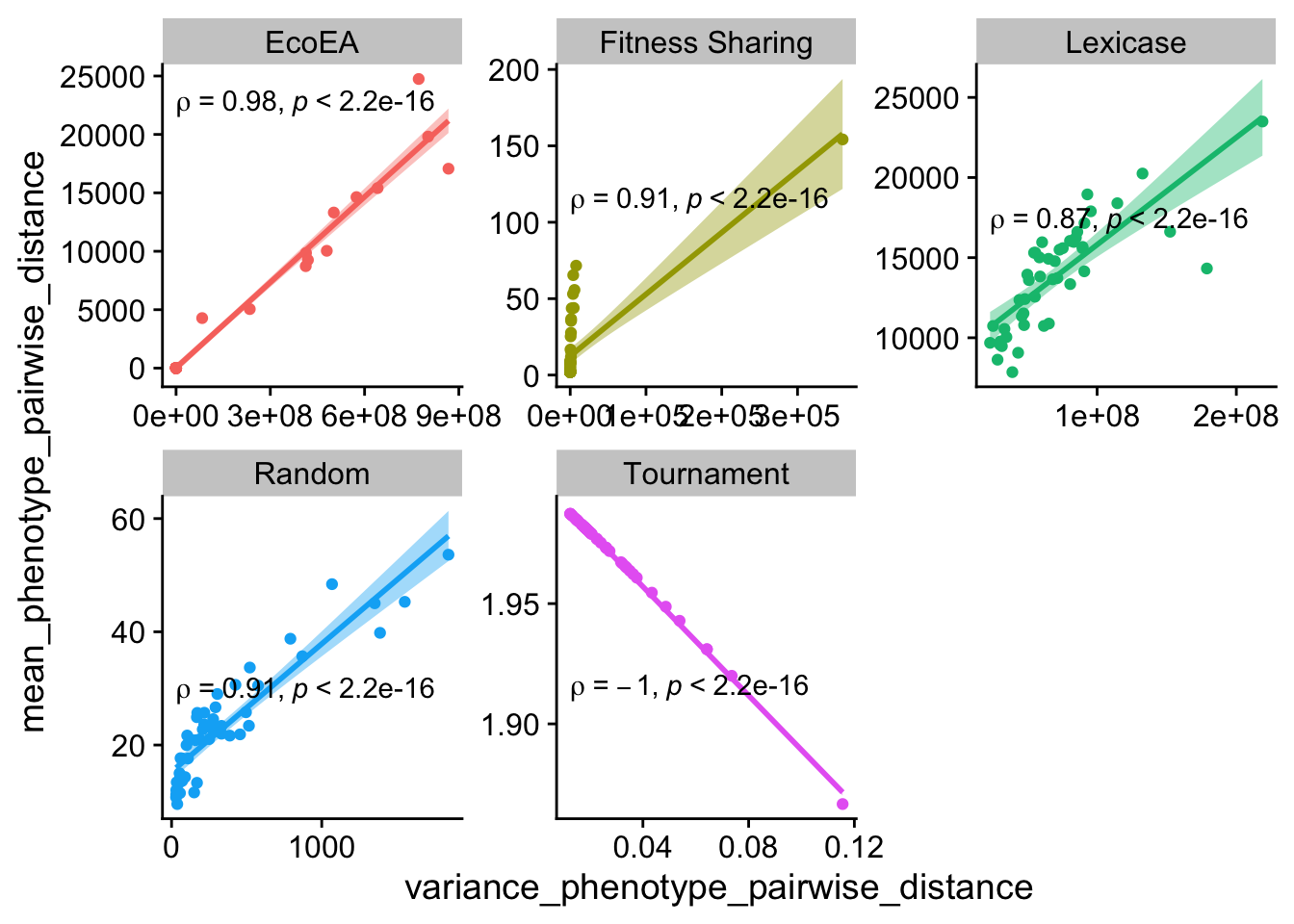
Mean and variance of pairwise distances appears to correlate fairly closely with each other across selection schemes. The exception to this is tournament selection, where the range of values for both of these metrics are very small and the correlation is directly inverse. This is likely being driven by there being a small number of taxa that are mostly siblings of each other.
ggplot(
data %>% filter(gen==500000),
aes(
y=mean_phenotype_pairwise_distance,
x=max_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")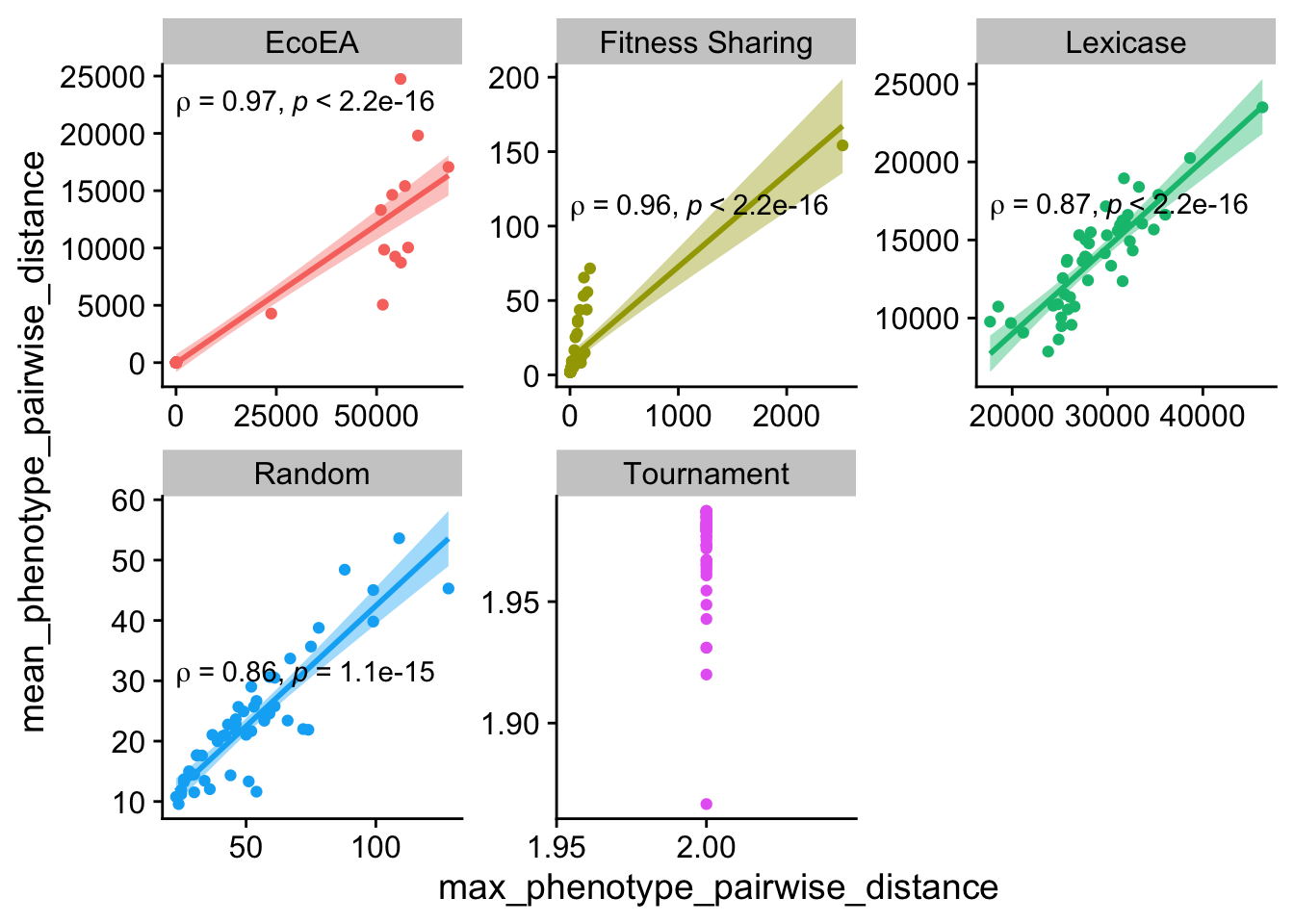
Maximum and mean pairwise distance also correlate pretty closely, with the exception again being tournament. These data shed further light on the previous graph as well - the maximum pariwise distance for tournament selection is always 2.
ggplot(
data %>% filter(gen==500000),
aes(
y=variance_phenotype_pairwise_distance,
x=max_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")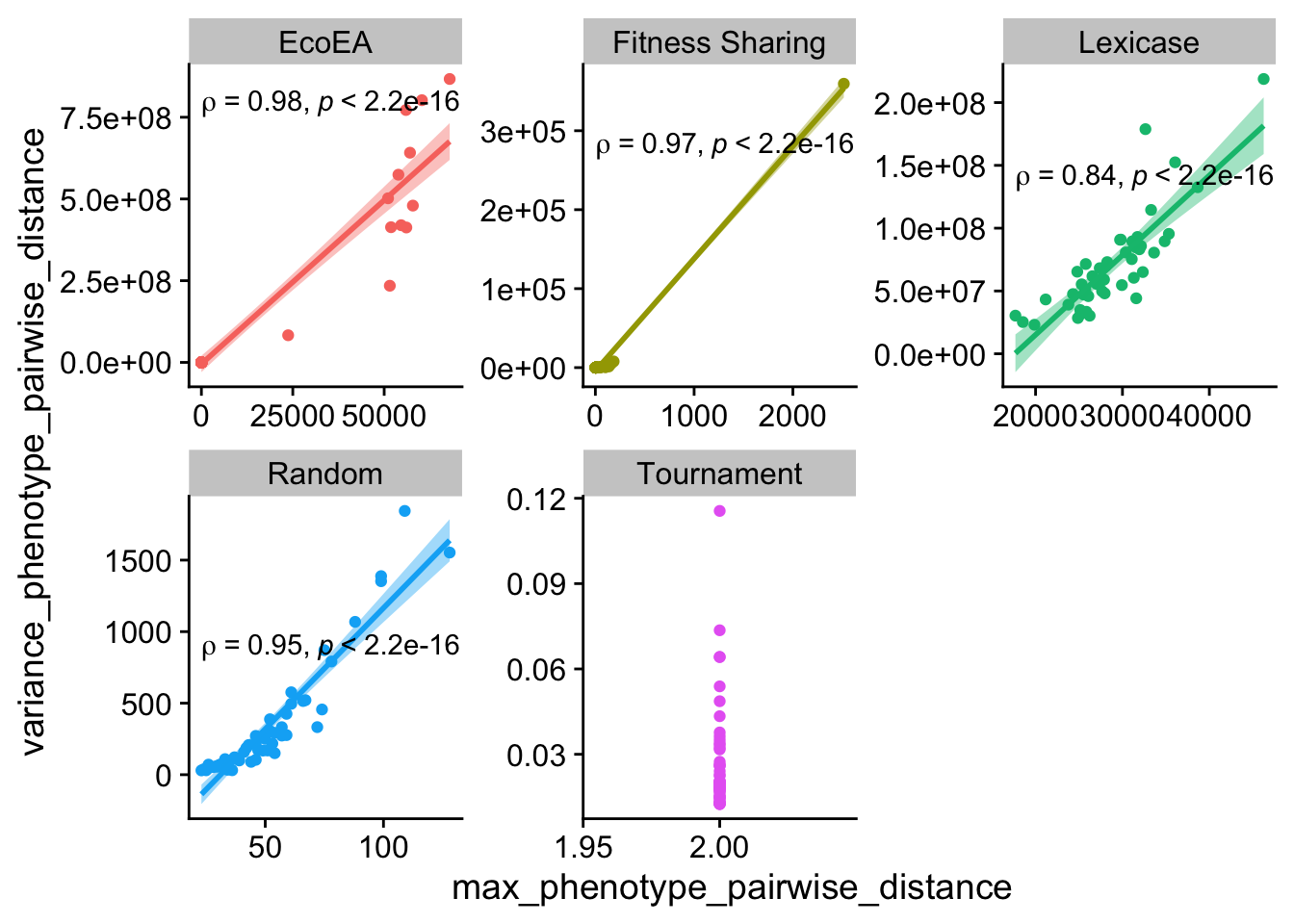
Max and variance of pairwise distances behave similarly.
And let’s check that it doesn’t look radically different early on in the run:
ggplot(
data %>% filter(gen==5000),
aes(
y=mean_phenotype_pairwise_distance,
x=variance_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")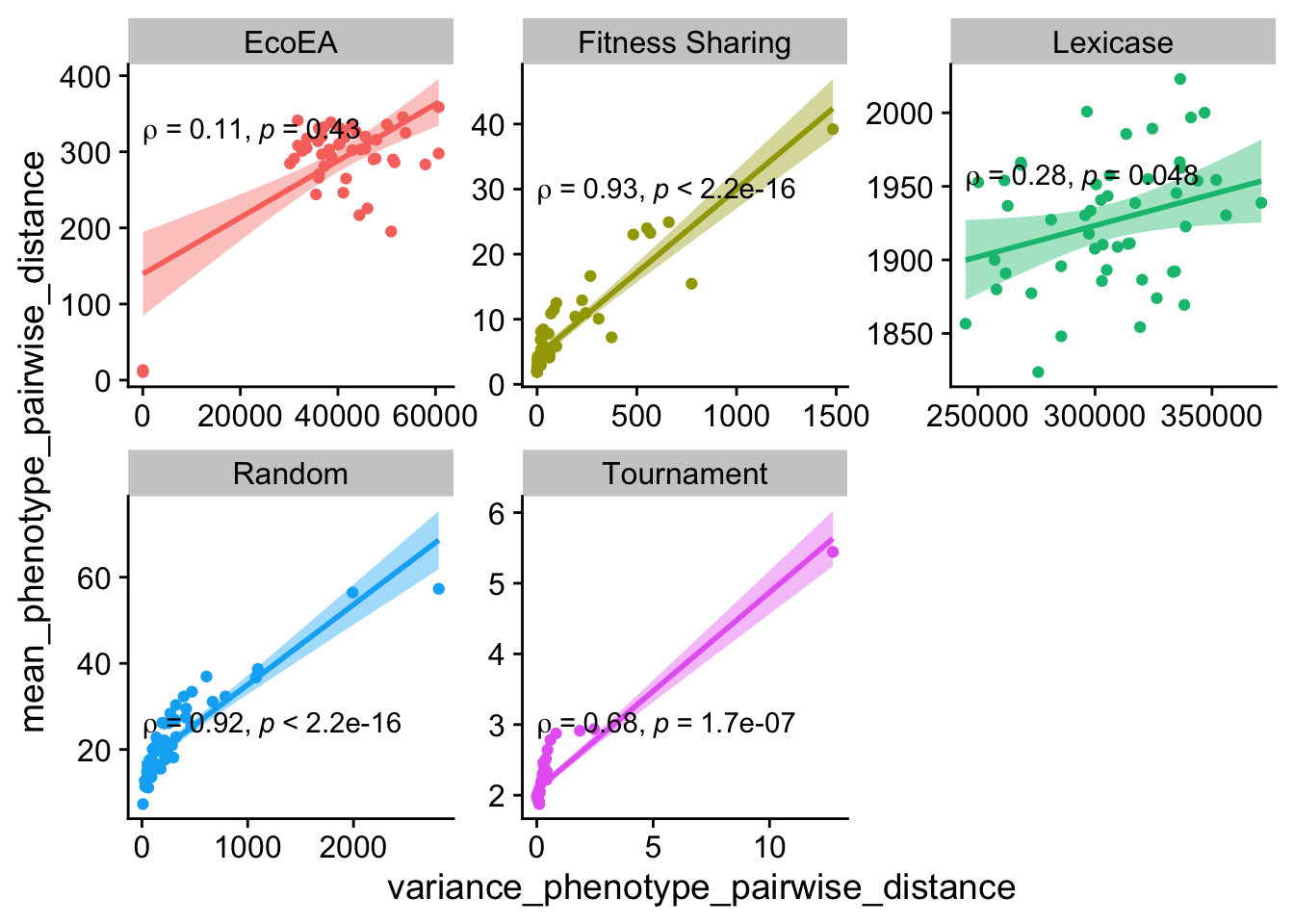
The correlation between these two metrics is substantially weaker for Eco-Ea and lexicase selection at the early time point.
ggplot(
data %>% filter(gen==5000),
aes(
y=mean_phenotype_pairwise_distance,
x=max_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")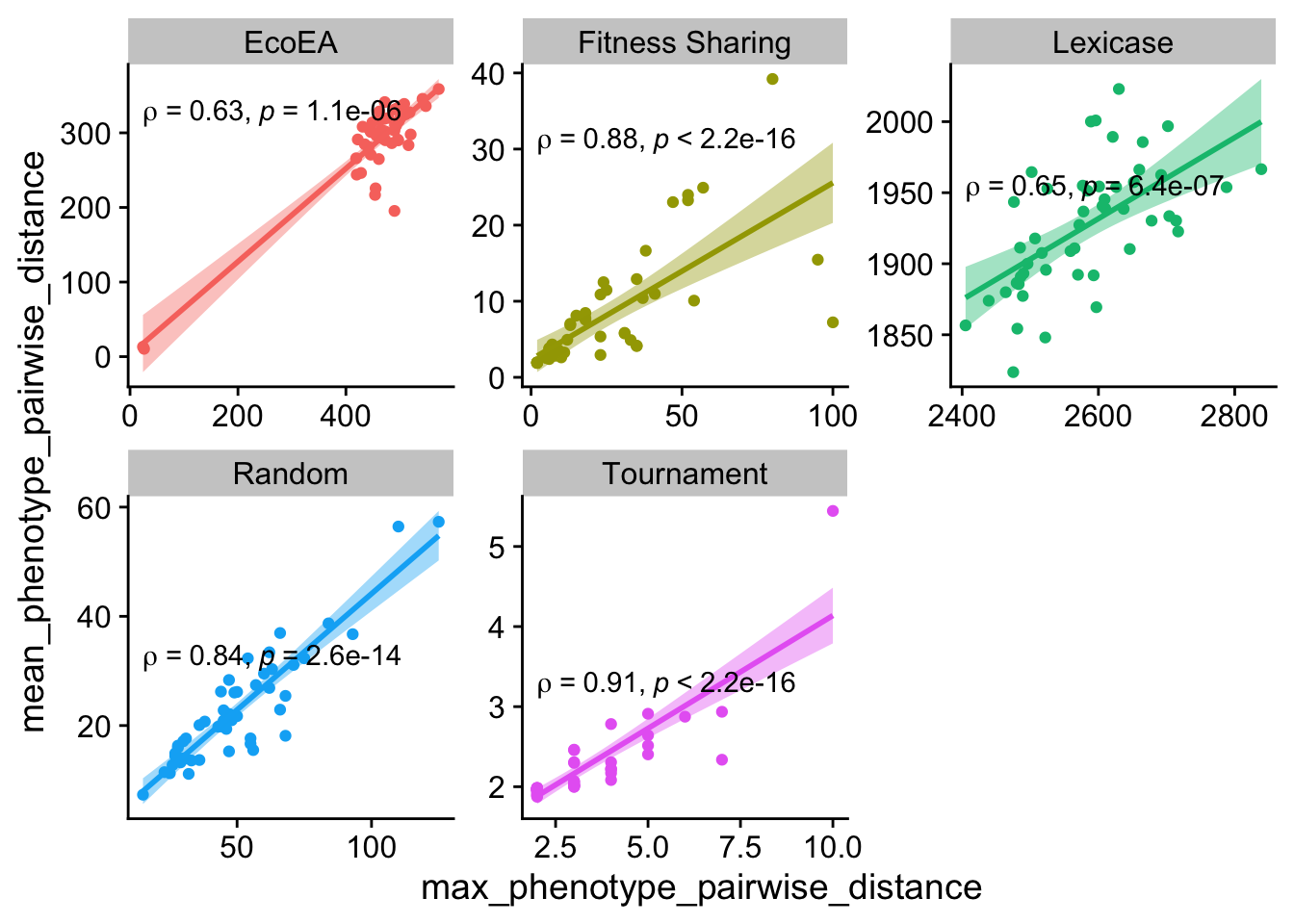
This relationship is also weaker, although not as much weaker.
ggplot(
data %>% filter(gen==5000),
aes(
y=variance_phenotype_pairwise_distance,
x=max_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")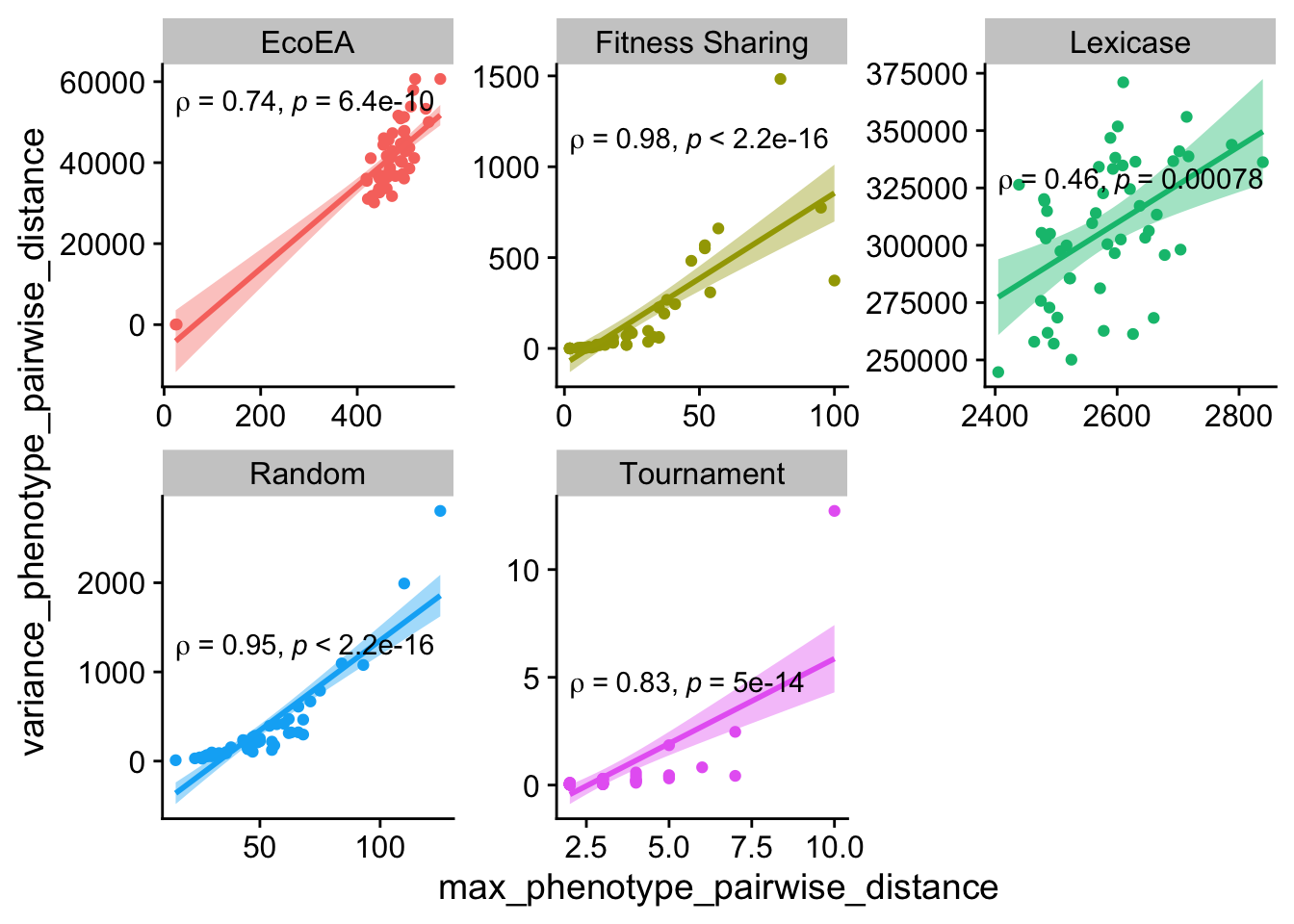
Similarly, the correlation between max and variance of pariwise distances is weakend, but still robust. So the relationship between mean and variance of pairwise distances is the only one that is really weaker at the earlier time points. Understanding exactly what drives these discrepancies is a promising angle for future work, as it may shed further light on the dynamics occurring in the run. It is particularly interesting that EcoEA and Lexicase selection are the scenarios where they diverge.
Similarly, the evolutionary distinctiveness metrics look largely similar to each other, but let’s spot check that too.
ggplot(
data %>% filter(gen==500000),
aes(
y=mean_phenotype_evolutionary_distinctiveness,
x=variance_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")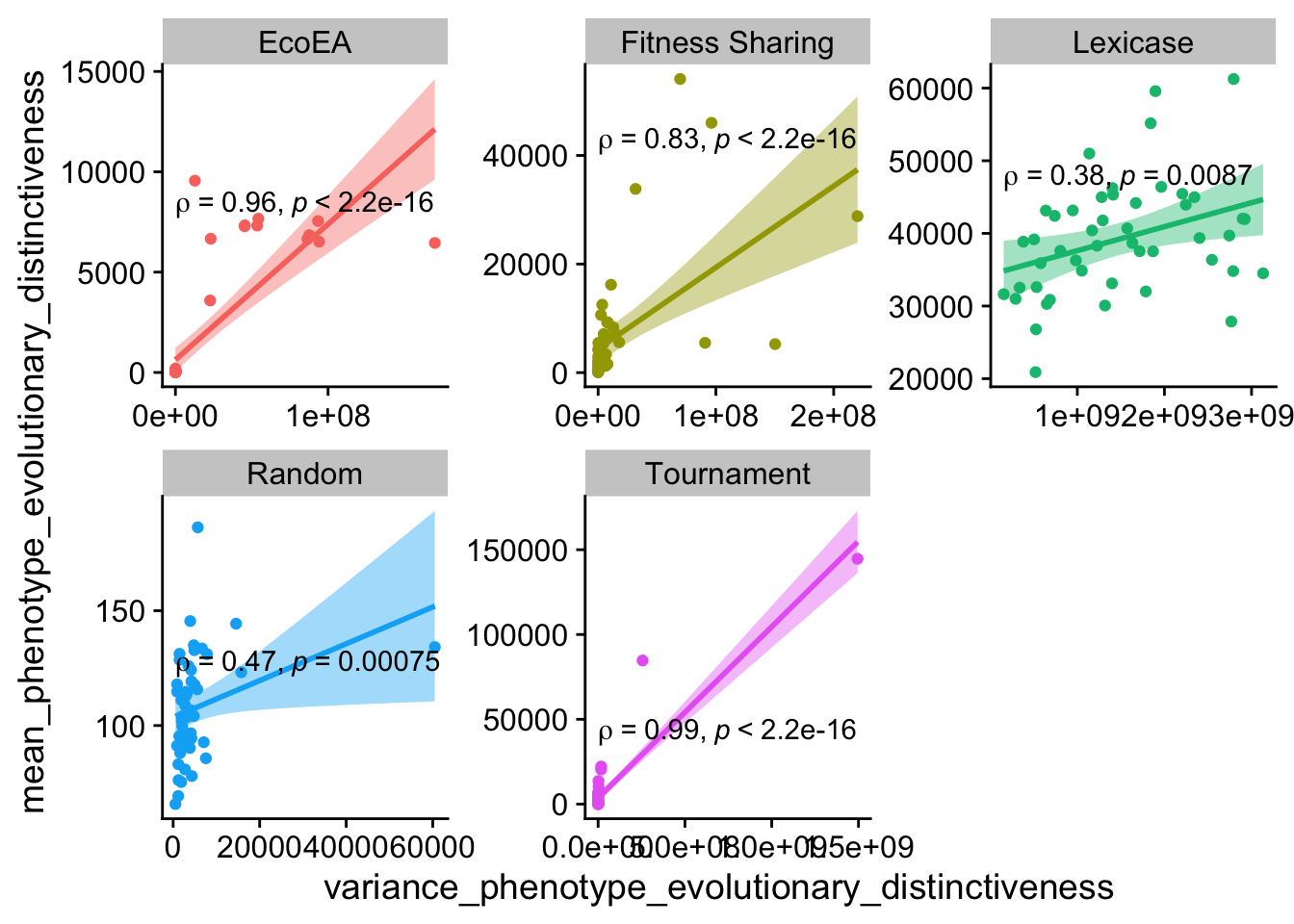
These aren’t the strongest correlations; different information is definitely being captured by each metric. However, they still correlate to a fair extent.
ggplot(
data %>% filter(gen==500000),
aes(
y=mean_phenotype_evolutionary_distinctiveness,
x=max_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")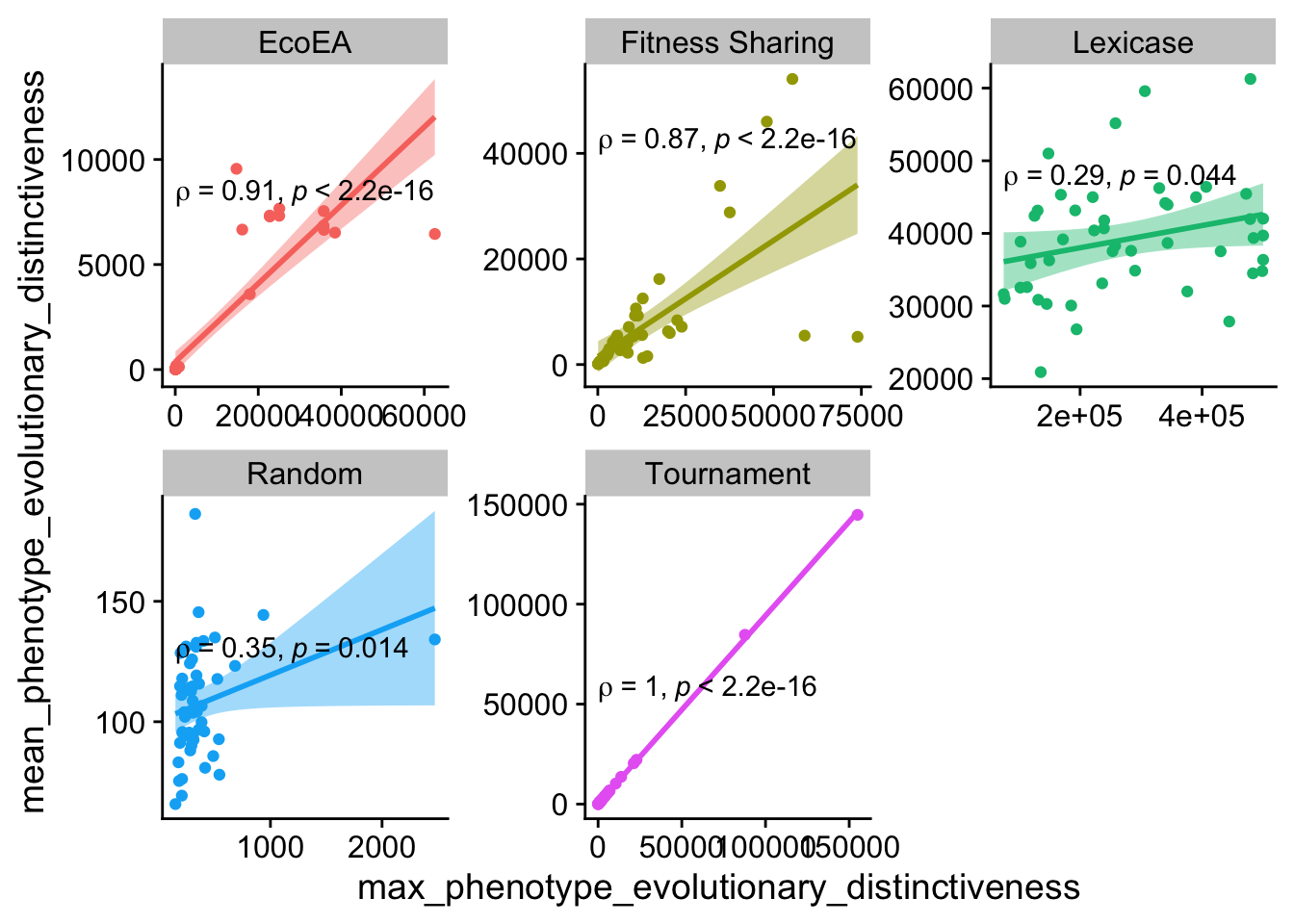
Again, for the most part there is a fairly high degree of correlation here.
ggplot(
data %>% filter(gen==500000),
aes(
y=mean_phenotype_evolutionary_distinctiveness,
x=min_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")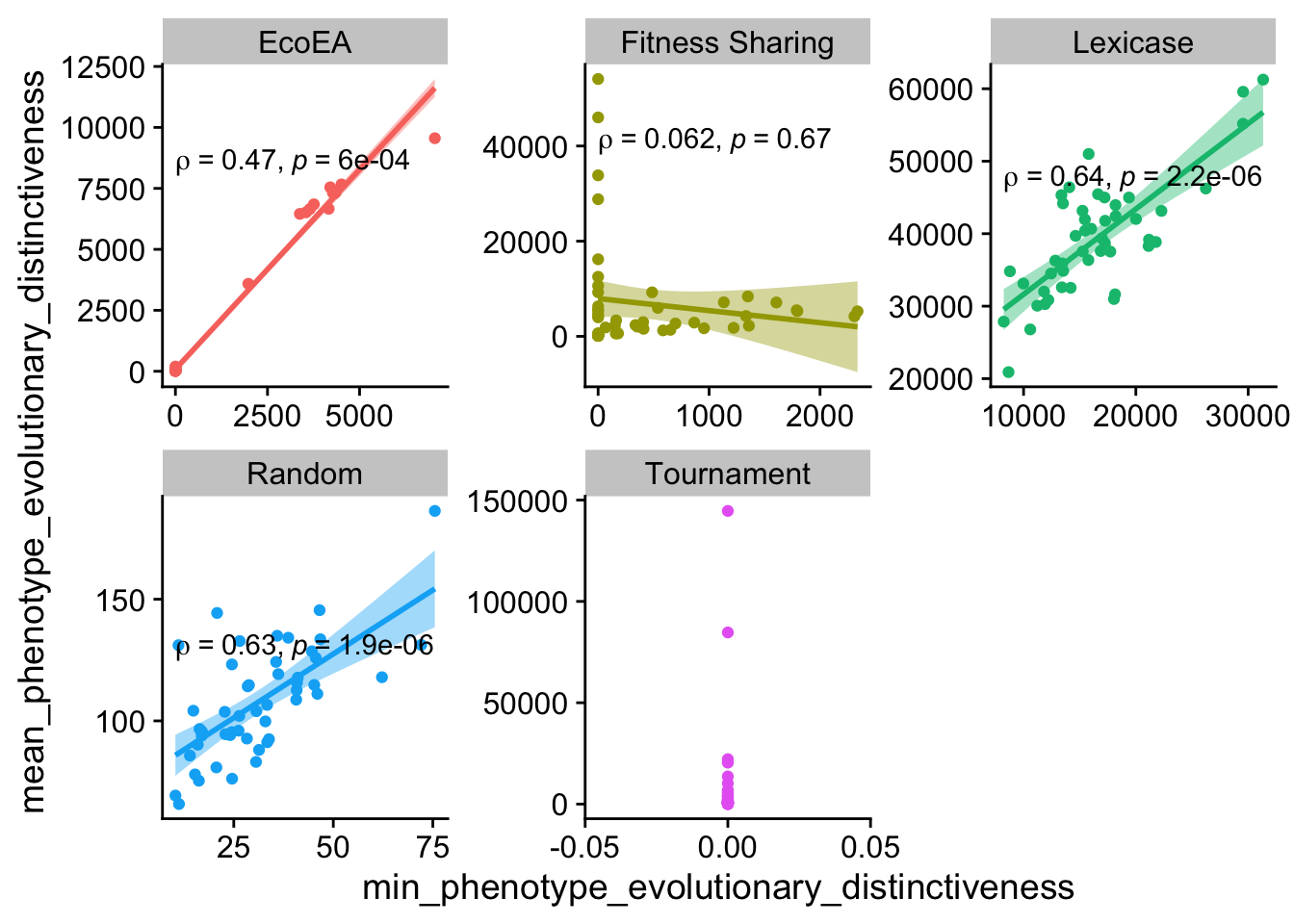
And here too, although it’s getting kind of weak for lexicase.
And let’s check out an earlier time point
ggplot(
data %>% filter(gen==5000),
aes(
y=mean_phenotype_evolutionary_distinctiveness,
x=variance_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")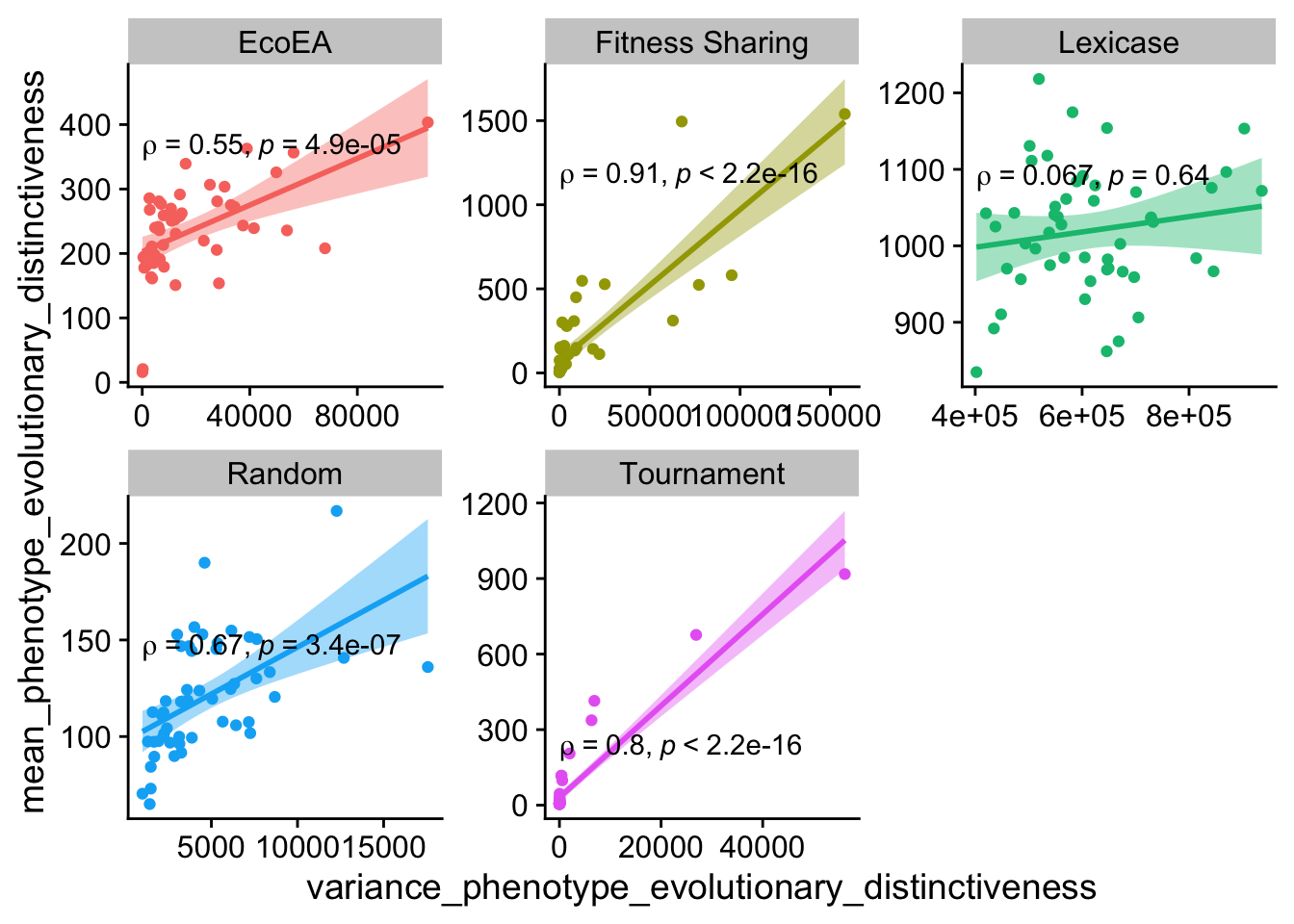
The relationship between min and mean evolutionary distinctiveness holds up at the earlier time point for everything except fitness sharing.
ggplot(
data %>% filter(gen==5000),
aes(
y=mean_phenotype_evolutionary_distinctiveness,
x=max_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")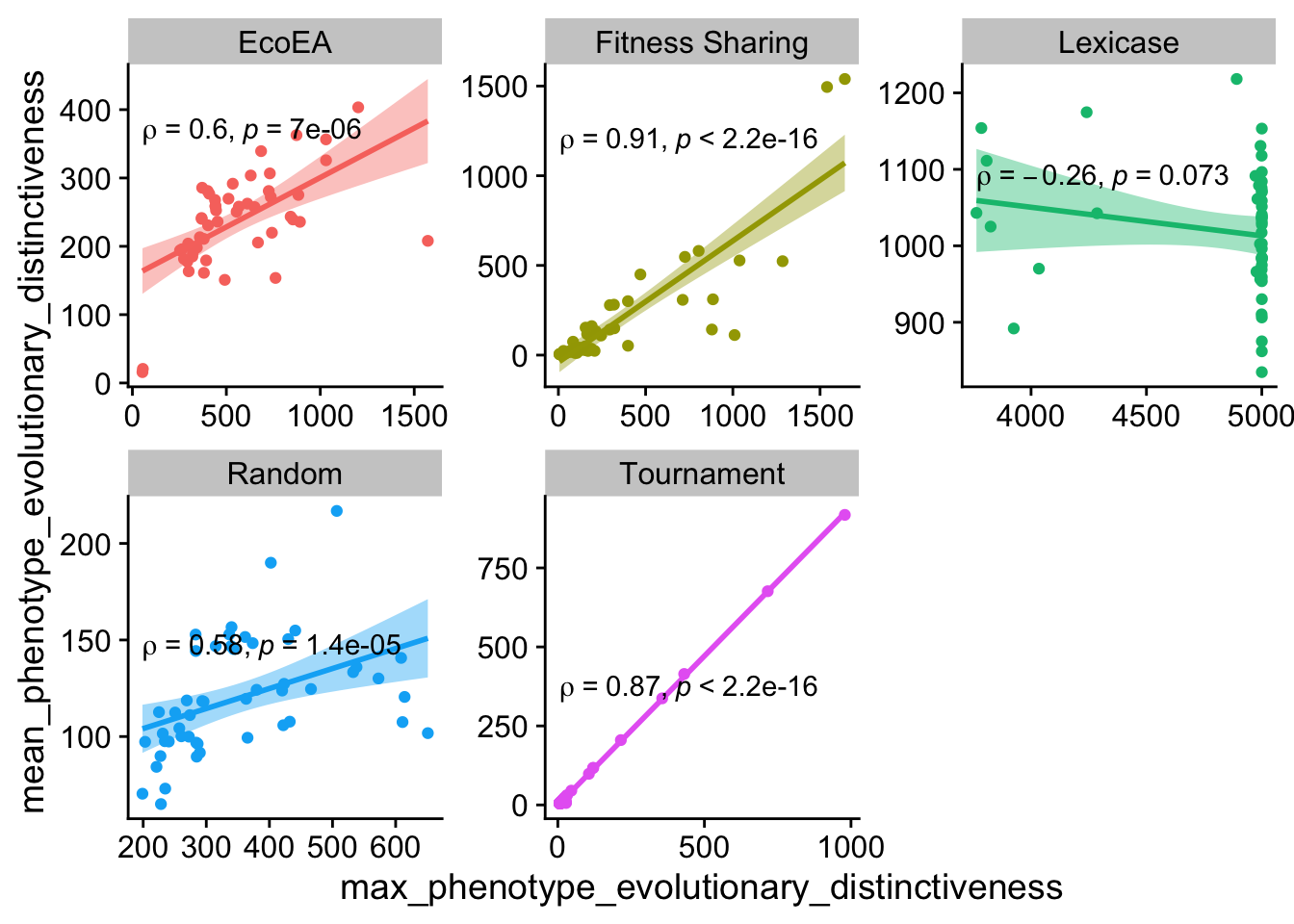
And here, for everything except lexicase
ggplot(
data %>% filter(gen==5000),
aes(
y=mean_phenotype_evolutionary_distinctiveness,
x=min_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_x_continuous(
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")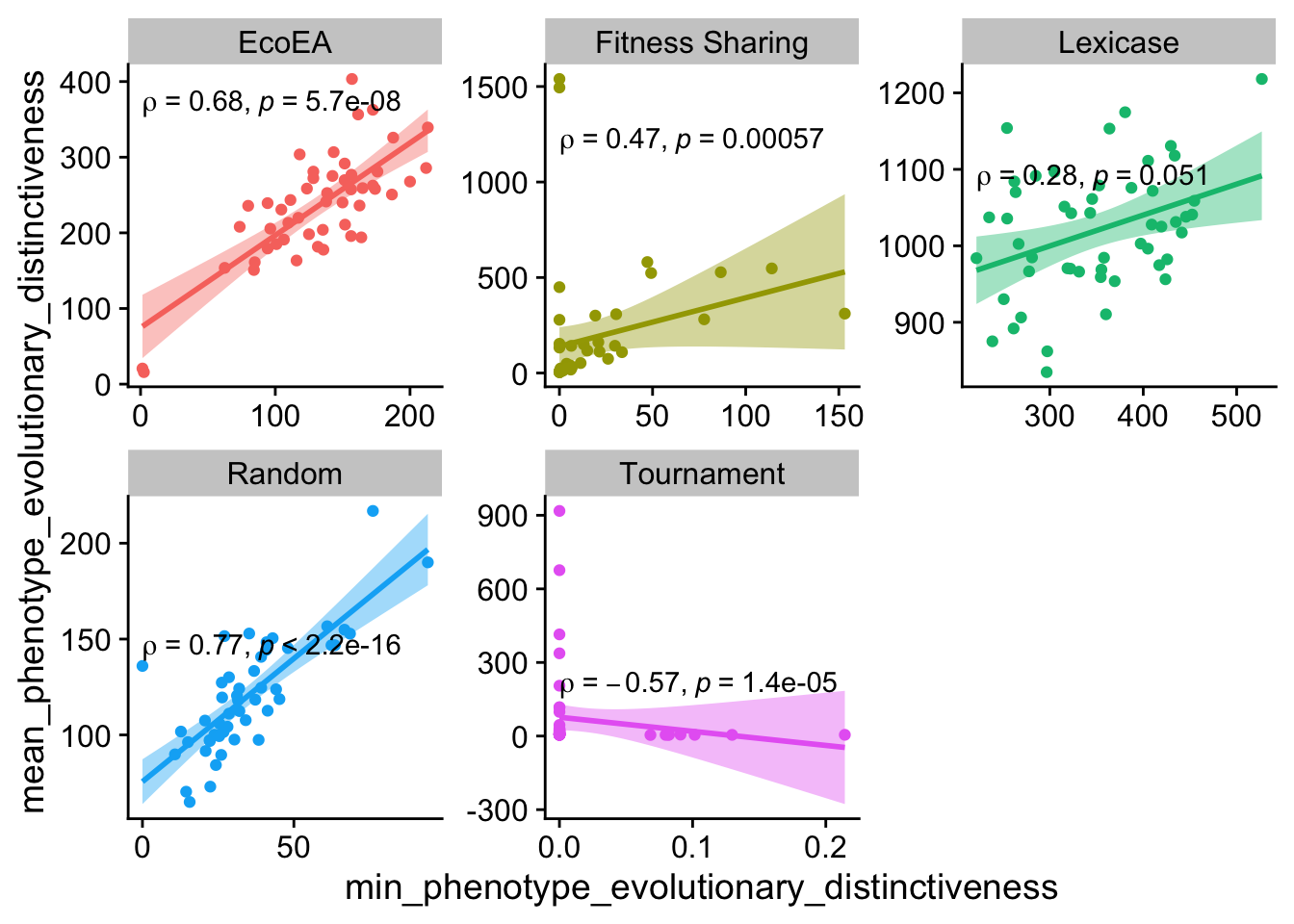
These are relatively weak for the most part.
Looks like the evolutionary distinctiveness statistics capture more different information from each other than the pairwise distance statistics (in general the correlations are weaker, and there are more deviations from the pattern of positive correlations). However, in a lot of cases they still correlate. There is definitely more exploration to be done on what differences in these metrics imply, but for simplicity we’ll stick to one pairwise distance metric and one evolutionary distinctiveness metric.
Since it’s hard to get a gestalt impression from the scatterplots, let’s take a look at the correlation matrices for each selection scheme. Note that these are at the final time point, which clearly misses some dynamics that occurr earlier.
2.3.1.1 Random
final_data %>%
filter(selection_name=="Random") %>%
transmute(MinPD=min_phenotype_pairwise_distance,
MeanPD=mean_phenotype_pairwise_distance,
MaxPD=max_phenotype_pairwise_distance,
VarPD=variance_phenotype_pairwise_distance,
MinED = min_phenotype_evolutionary_distinctiveness,
MeanED= mean_phenotype_evolutionary_distinctiveness,
MaxED=max_phenotype_evolutionary_distinctiveness,
VarED=variance_phenotype_evolutionary_distinctiveness,
PD=phenotype_current_phylogenetic_diversity, # See Faith 1992
MRCA=phen_mrca_depth, # Phylogenetic depth of most recent common ancestor
N=phen_num_taxa # Number of taxonomically-distinct phenotypes
) %>%
cor_mat() %>%
pull_lower_triangle() %>%
cor_plot()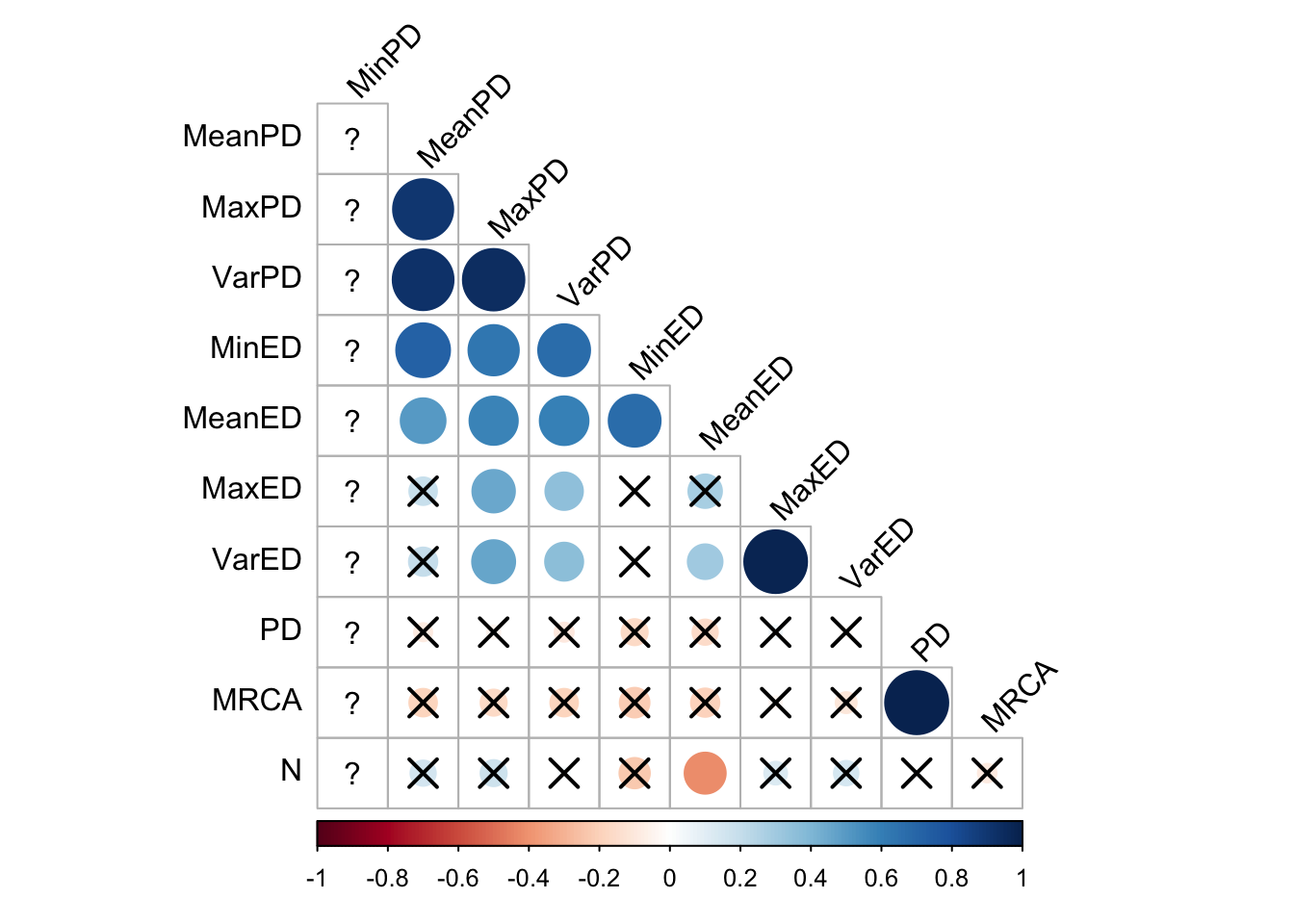
In random selection, all the pairwise distance metrics correlate with each other, and also more weakly with some of the evolutionary distinctiveness metrics, but not max and variance (although they correlate strongly with each other). It is somewhat surprising that the depth of the most recent common ancestor and Faith’s Phylogenetic Diveristy metric have so little correlation with the others.
2.3.1.2 Tournament
final_data %>%
filter(selection_name=="Tournament") %>%
transmute(MinPD=min_phenotype_pairwise_distance,
MeanPD=mean_phenotype_pairwise_distance,
MaxPD=max_phenotype_pairwise_distance,
VarPD=variance_phenotype_pairwise_distance,
MinED = min_phenotype_evolutionary_distinctiveness,
MeanED= mean_phenotype_evolutionary_distinctiveness,
MaxED=max_phenotype_evolutionary_distinctiveness,
VarED=variance_phenotype_evolutionary_distinctiveness,
PD=phenotype_current_phylogenetic_diversity, # See Faith 1992
MRCA=phen_mrca_depth, # Phylogenetic depth of most recent common ancestor
N=phen_num_taxa # Number of taxonomically-distinct phenotypes
) %>%
cor_mat() %>%
pull_lower_triangle() %>%
cor_plot()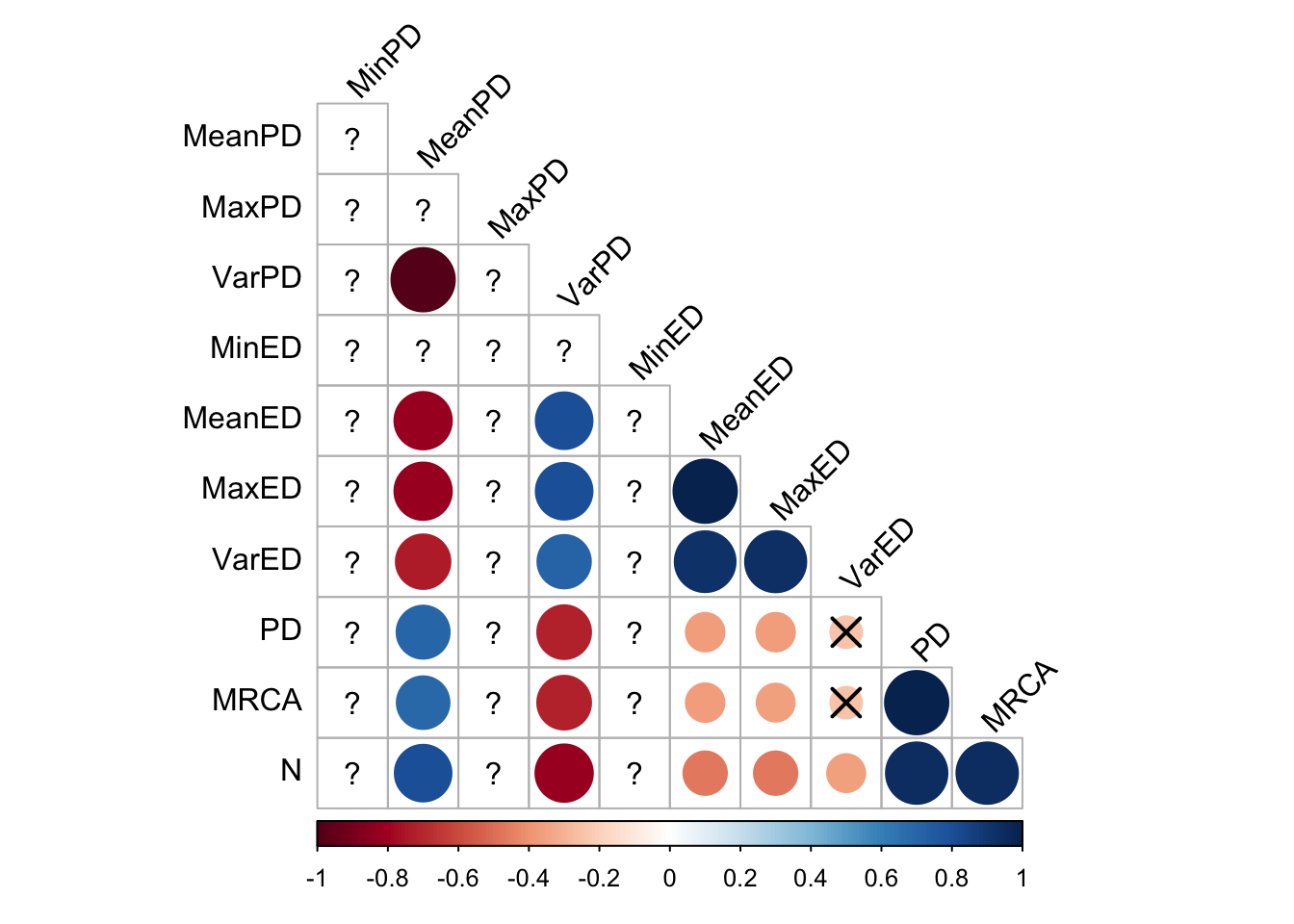
Here, all the evolutionary distinctiveness metrics coreelate strongly with each other, but the pairiwse distance metrics less so (although this is likely an artifact of the extremely low phylogenetic diversity in tournament selection)
2.3.1.3 Fitness Sharing
final_data %>%
filter(selection_name=="Fitness Sharing") %>%
transmute(MinPD=min_phenotype_pairwise_distance,
MeanPD=mean_phenotype_pairwise_distance,
MaxPD=max_phenotype_pairwise_distance,
VarPD=variance_phenotype_pairwise_distance,
MinED = min_phenotype_evolutionary_distinctiveness,
MeanED= mean_phenotype_evolutionary_distinctiveness,
MaxED=max_phenotype_evolutionary_distinctiveness,
VarED=variance_phenotype_evolutionary_distinctiveness,
PD=phenotype_current_phylogenetic_diversity, # See Faith 1992
MRCA=phen_mrca_depth, # Phylogenetic depth of most recent common ancestor
N=phen_num_taxa # Number of taxonomically-distinct phenotypes
) %>%
cor_mat() %>%
pull_lower_triangle() %>%
cor_plot()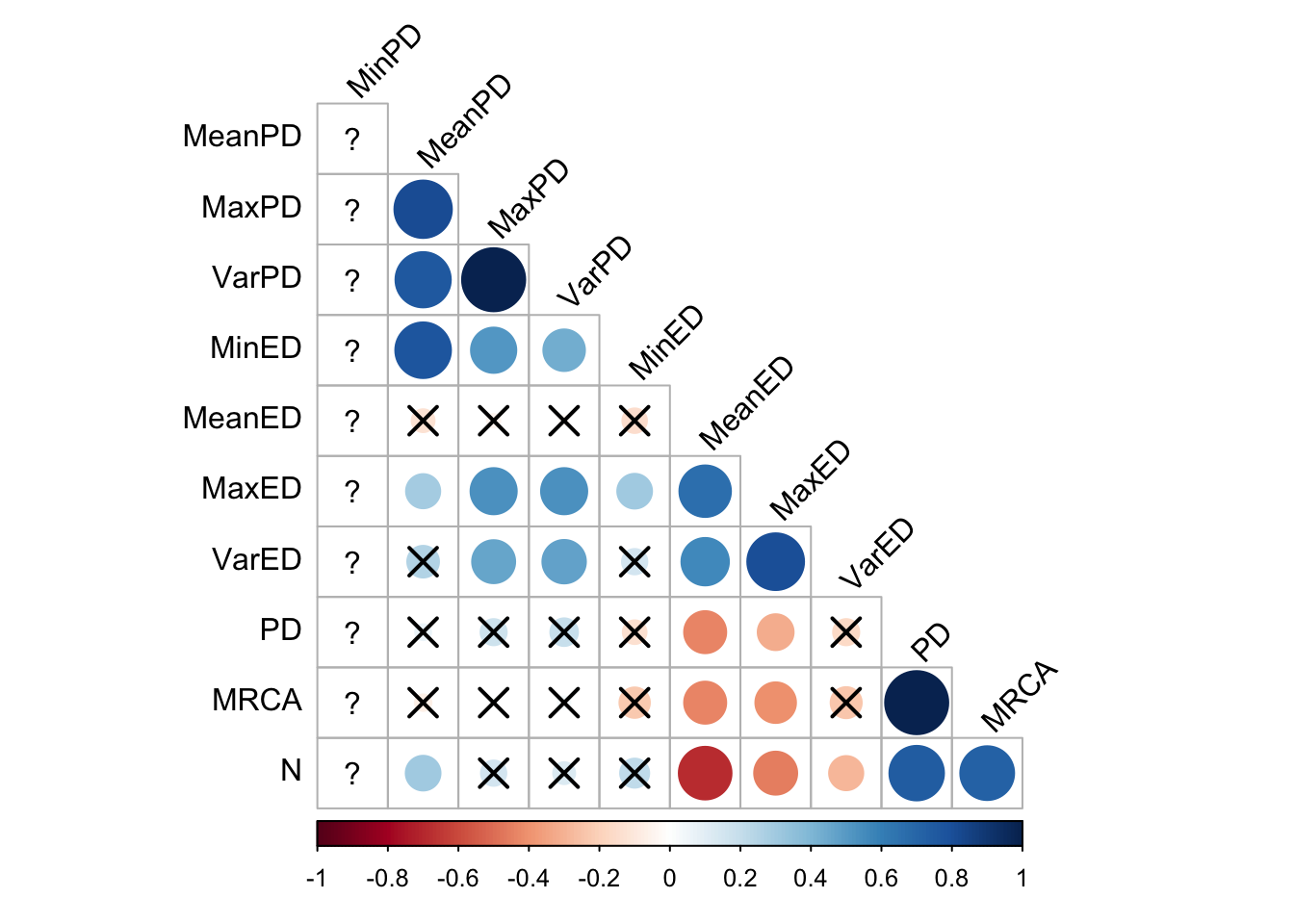
Here, the pairwise distance metrics correlate with each other and the evolutionary distinctivenes metrics correlated with each other. There are some correlations between pairwise distance and evolutionary distinctiveness metrics, but they’re weaker.
2.3.1.4 Lexicase selection
final_data %>%
filter(selection_name=="Lexicase") %>%
transmute(MinPD=min_phenotype_pairwise_distance,
MeanPD=mean_phenotype_pairwise_distance,
MaxPD=max_phenotype_pairwise_distance,
VarPD=variance_phenotype_pairwise_distance,
MinED = min_phenotype_evolutionary_distinctiveness,
MeanED= mean_phenotype_evolutionary_distinctiveness,
MaxED=max_phenotype_evolutionary_distinctiveness,
VarED=variance_phenotype_evolutionary_distinctiveness,
PD=phenotype_current_phylogenetic_diversity, # See Faith 1992
MRCA=phen_mrca_depth, # Phylogenetic depth of most recent common ancestor
N=phen_num_taxa # Number of taxonomically-distinct phenotypes
) %>%
cor_mat() %>%
pull_lower_triangle() %>%
cor_plot()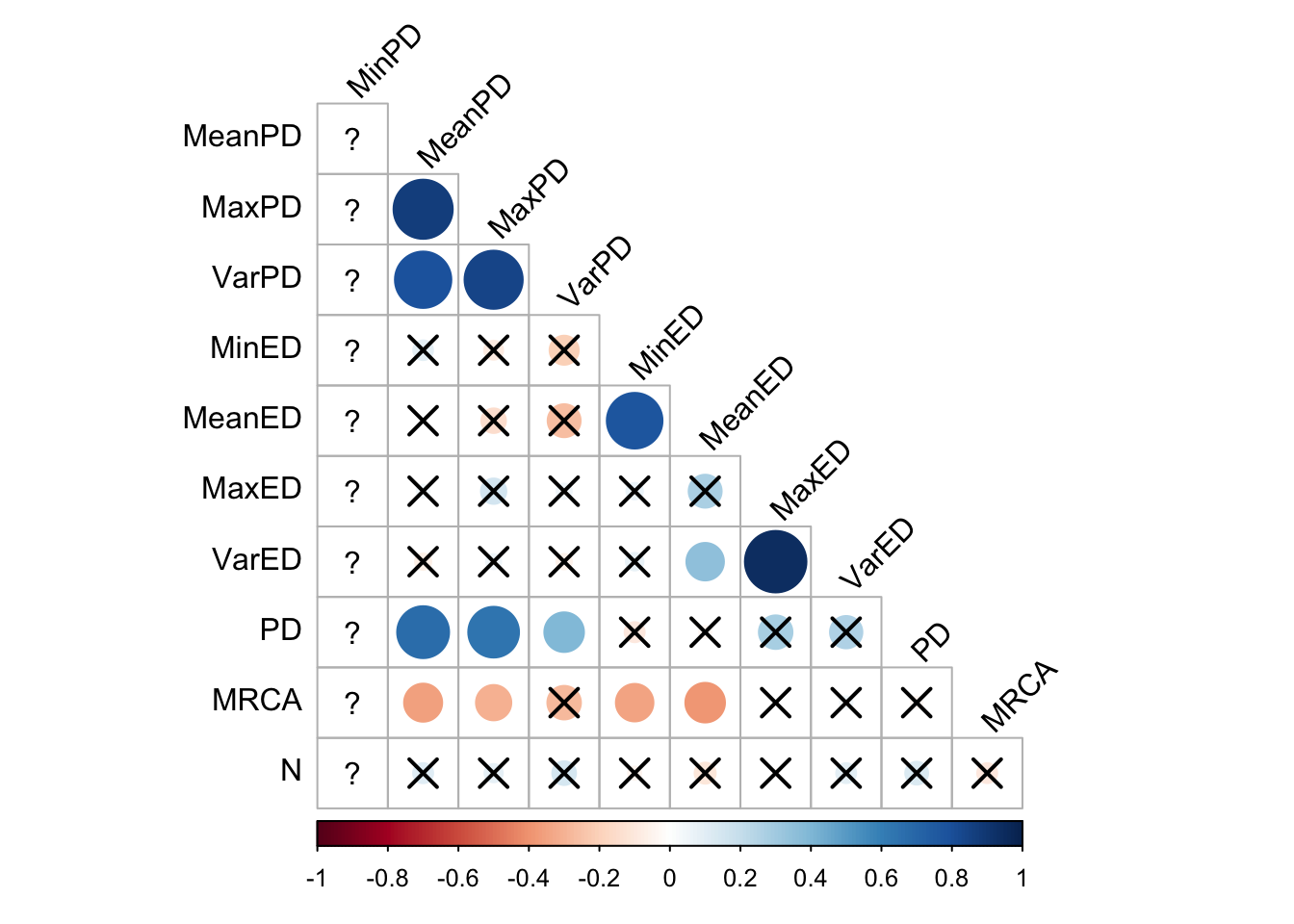
Again, the pairwise distance metrics correlate with each other but not wth the volutionary distinctivness metrics. The evolutionary distinctiveness metrics sort of correlate with each other.
2.3.1.5 Eco-EA
final_data %>%
filter(selection_name=="EcoEA") %>%
transmute(MinPD=min_phenotype_pairwise_distance,
MeanPD=mean_phenotype_pairwise_distance,
MaxPD=max_phenotype_pairwise_distance,
VarPD=variance_phenotype_pairwise_distance,
MinED = min_phenotype_evolutionary_distinctiveness,
MeanED= mean_phenotype_evolutionary_distinctiveness,
MaxED=max_phenotype_evolutionary_distinctiveness,
VarED=variance_phenotype_evolutionary_distinctiveness,
PD=phenotype_current_phylogenetic_diversity, # See Faith 1992
MRCA=phen_mrca_depth, # Phylogenetic depth of most recent common ancestor
N=phen_num_taxa # Number of taxonomically-distinct phenotypes
) %>%
cor_mat() %>%
pull_lower_triangle() %>%
cor_plot()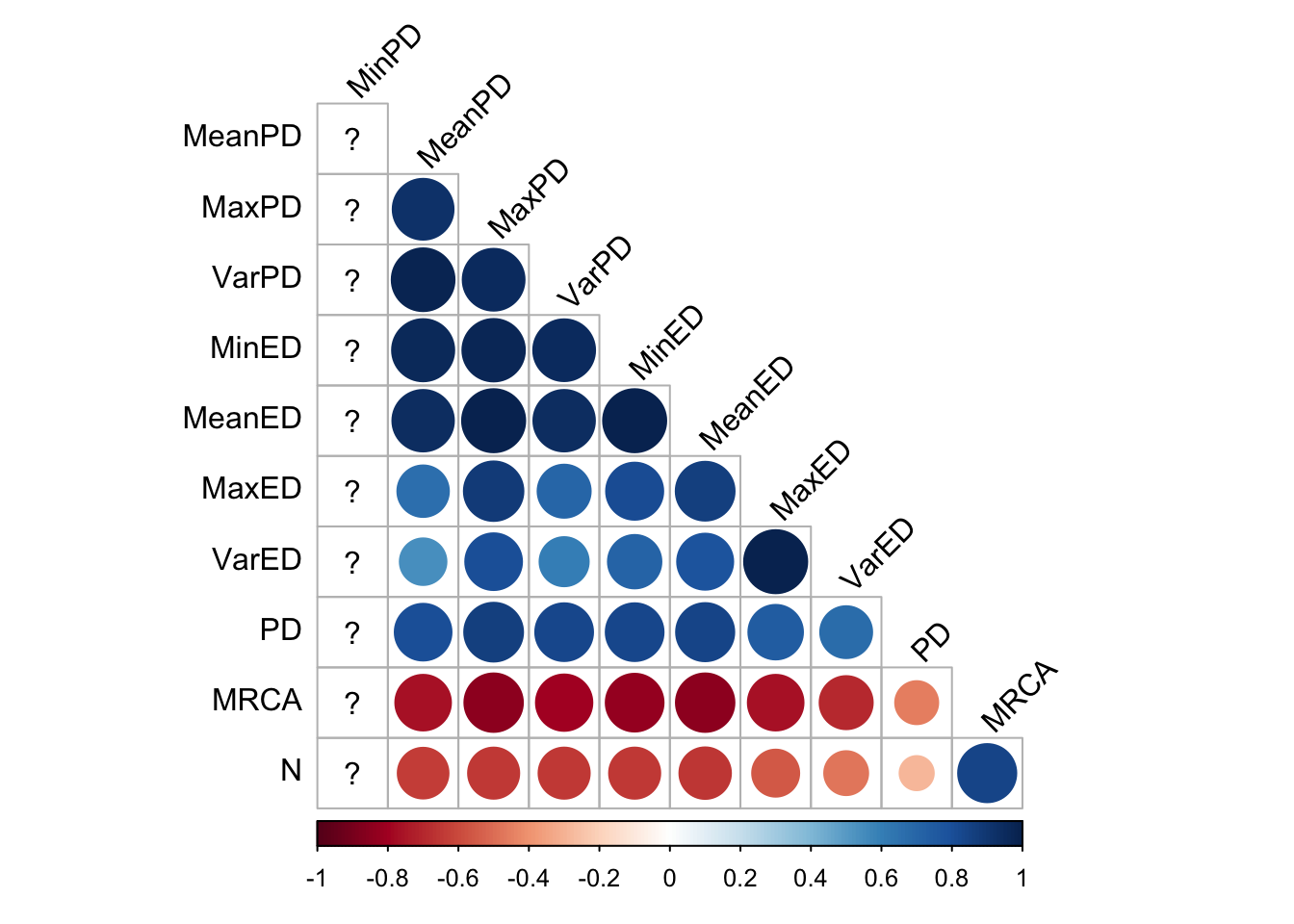
In Eco-EA, there is an impressively strong positive correlation among all the phylodiversity metrics. Understanding what causes Eco-EA to deviate from lexicase selection (and to a lesser extent fitness sharing) in this way would be worthy of further research.
2.3.2 Over time
2.3.2.1 Mean pairwise distance
First we plot mean pairwise distance over time. We log the y axis because there is such variation in mean pairwise distance across selection schemes, and the x-axis for the same reason as before.
ggplot(
data,
aes(
x=gen,
y=mean_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
stat_summary(geom="line", fun=mean) +
stat_summary(
geom="ribbon",
fun.data="mean_cl_boot",
fun.args=list(conf.int=0.95),
alpha=0.2,
linetype=0
) +
scale_y_log10(
name="Mean pairwise distance"
) +
scale_x_log10(
name="Generation"
) +
scale_color_discrete("Selection") +
scale_fill_discrete("Selection")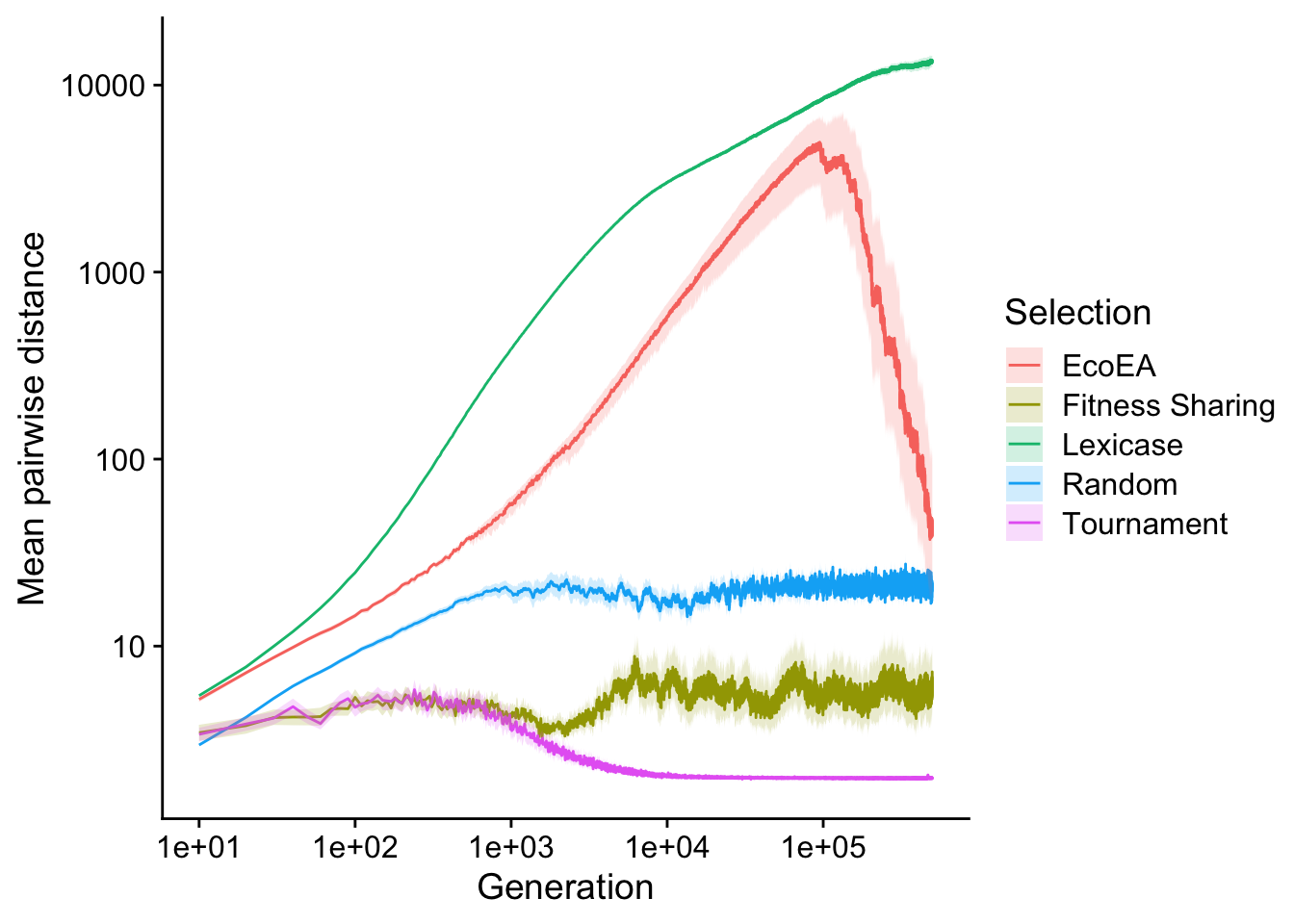
Lexicase selection maintains a monotonic increase in phylogenetic diversity over the course of the entire experiment. It likely never experiences a full coalescence event (where the most-recent common ancestor changes). Eco-EA nearly keeps pace with lexicase selection until towards the end, when its phylogenetic diversity crashes. This is likely the result of selective sweeps that begin to occur as the population discovers high fitness solutions. Fitness sharing shows a slight dip at the same time that its fitness plateaus (likely also the result of a selective sweep), but phylogenetic diversity recovers afterwards, making for a relatively constant level. over time. Tournament selection, on the other hand, maintains the same (low) level of phylogenetic diversity as fitness sharing, up until the point that fitness plateaus, at which point tournament selection’s phylodiversity drops to nearly 0. Interestingly, lexicase selection and Eco-EA both maintain more phylodiversity than random selection, whereas fitness sharing and tournament selection maintain less.
2.3.2.2 Mean evolutionary distinctiveness
For comparison, we make the same plot with mean evolutionary distinctiveness.
ggplot(
data,
aes(
x=gen,
y=mean_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
stat_summary(geom="line", fun=mean) +
stat_summary(
geom="ribbon",
fun.data="mean_cl_boot",
fun.args=list(conf.int=0.95),
alpha=0.2,
linetype=0
) +
scale_y_log10(
name="Mean evolutionary distinctiveness"
) +
scale_x_log10(
name="Generation"
) +
scale_color_discrete("Selection") +
scale_fill_discrete("Selection")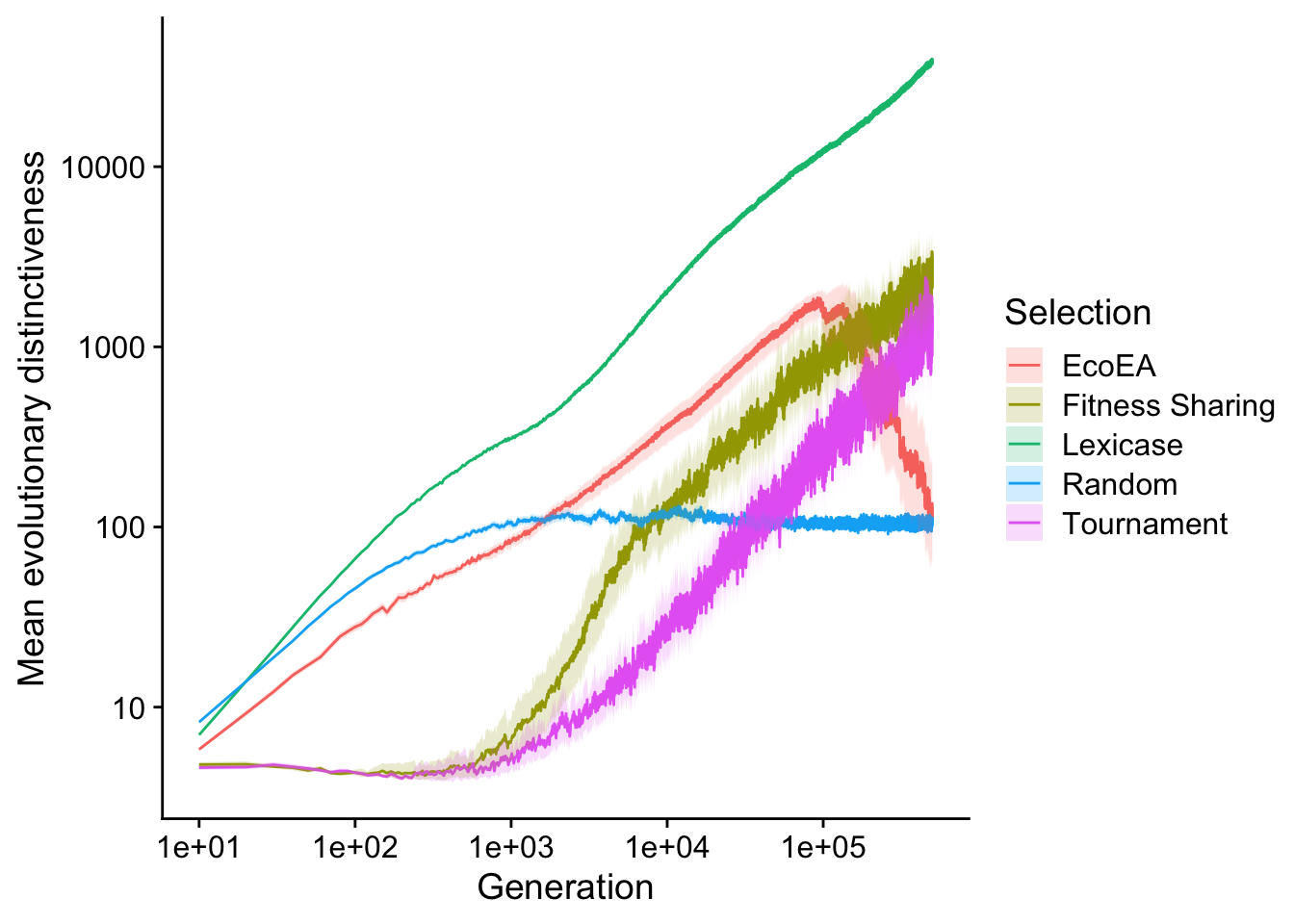
Interestingly, Fitness Sharing and tournament selection both start increasing in evolutionary distinctiveness only after their fitnesses have plateaued. This seems likely to be due to some sort of pathological behavior of mean evolutionary distinctiveness on small trees, but more investigation would be necessary to figure out exactly what’s going on. Trends in other selection schemes are largely similar.
2.3.3 Final
Next, we perform a more in-depth analysis of phylodiversity distributions at the final time point.
2.3.3.1 Mean pairwise distance
First, we test which selection schemes end up with significantly different final distributions of mean pairwise distance.
# Pairwise wilcoxon teset to determine which conditions are significantly different from each other
stat.test <- final_data %>%
wilcox_test(mean_phenotype_pairwise_distance ~ selection_name) %>%
adjust_pvalue(method = "bonferroni") %>%
add_significance() %>%
add_xy_position(x="selection_name",step.increase=1)
stat.test$label <- mapply(p_label,stat.test$p.adj)
# Output stats
stat.test %>%
kbl() %>%
kable_styling(
bootstrap_options = c(
"striped",
"hover",
"condensed",
"responsive"
)
) %>%
scroll_box(width="600px")| .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | y.position | groups | xmin | xmax | label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mean_phenotype_pairwise_distance | EcoEA | Fitness Sharing | 50 | 50 | 1824.0 | 7.70e-05 | 7.70e-04 | *** | 49488.81 | EcoEA , Fitness Sharing | 1 | 2 | p = 0.00077 |
| mean_phenotype_pairwise_distance | EcoEA | Lexicase | 50 | 47 | 227.0 | 0.00e+00 | 0.00e+00 | **** | 76981.49 | EcoEA , Lexicase | 1 | 3 | p < 1e-04 |
| mean_phenotype_pairwise_distance | EcoEA | Random | 50 | 50 | 690.0 | 1.15e-04 | 1.15e-03 | ** | 104474.18 | EcoEA , Random | 1 | 4 | p = 0.00115 |
| mean_phenotype_pairwise_distance | EcoEA | Tournament | 50 | 50 | 2500.0 | 0.00e+00 | 0.00e+00 | **** | 131966.86 | EcoEA , Tournament | 1 | 5 | p < 1e-04 |
| mean_phenotype_pairwise_distance | Fitness Sharing | Lexicase | 50 | 47 | 0.0 | 0.00e+00 | 0.00e+00 | **** | 159459.54 | Fitness Sharing, Lexicase | 2 | 3 | p < 1e-04 |
| mean_phenotype_pairwise_distance | Fitness Sharing | Random | 50 | 50 | 536.0 | 9.00e-07 | 8.70e-06 | **** | 186952.22 | Fitness Sharing, Random | 2 | 4 | p < 1e-04 |
| mean_phenotype_pairwise_distance | Fitness Sharing | Tournament | 50 | 50 | 2232.5 | 0.00e+00 | 0.00e+00 | **** | 214444.90 | Fitness Sharing, Tournament | 2 | 5 | p < 1e-04 |
| mean_phenotype_pairwise_distance | Lexicase | Random | 47 | 50 | 2350.0 | 0.00e+00 | 0.00e+00 | **** | 241937.58 | Lexicase, Random | 3 | 4 | p < 1e-04 |
| mean_phenotype_pairwise_distance | Lexicase | Tournament | 47 | 50 | 2350.0 | 0.00e+00 | 0.00e+00 | **** | 269430.26 | Lexicase , Tournament | 3 | 5 | p < 1e-04 |
| mean_phenotype_pairwise_distance | Random | Tournament | 50 | 50 | 2500.0 | 0.00e+00 | 0.00e+00 | **** | 296922.94 | Random , Tournament | 4 | 5 | p < 1e-04 |
Looks like they are all significantly different from each other.
# Raincloud plot of final mean pairwise distance
final_phylogeny_fig <- ggplot(
final_data,
aes(
x=selection_name,
y=mean_phenotype_pairwise_distance,
fill=selection_name
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
scale="width"
) +
geom_point(
mapping=aes(color=selection_name),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_log10(
name="Mean pairwise distance"
) +
scale_x_discrete(
name="Selection"
) +
scale_fill_discrete(
name="Selection"
) +
scale_color_discrete(
name="Selection"
) +
theme(legend.position = "none")
final_phylogeny_fig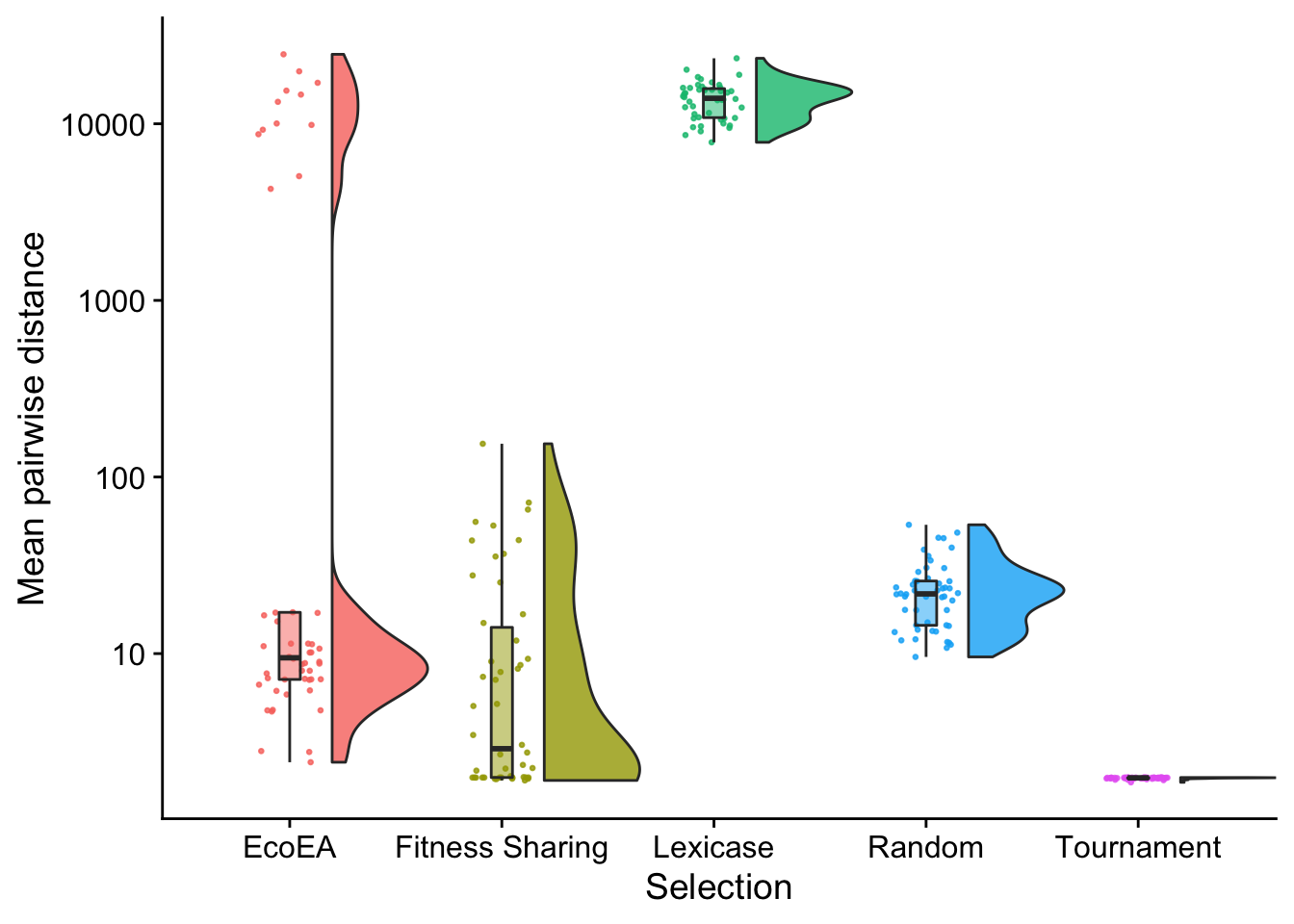
This shows something interesting! Final phylogenetic diversity in Eco-EA is heavily bimodal. In later analysis, we will see that the runs with high phylogenetic diversity are the ones with lower fitness, suggesting that they have no yet experienced a selective sweep resulting from the discovery of a high-fitness solution.
2.3.3.2 Mean evolutionary distinctiveness
Tests for significant differences:
# Pairwise wilcoxon teset to determine which conditions are significantly different from each other
stat.test <- final_data %>%
wilcox_test(mean_phenotype_evolutionary_distinctiveness ~ selection_name) %>%
adjust_pvalue(method = "bonferroni") %>%
add_significance() %>%
add_xy_position(x="selection_name",step.increase=1)
stat.test$label <- mapply(p_label,stat.test$p.adj)
# Output stats
stat.test %>%
kbl() %>%
kable_styling(
bootstrap_options = c(
"striped",
"hover",
"condensed",
"responsive"
)
) %>%
scroll_box(width="100%")| .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | y.position | groups | xmin | xmax | label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mean_phenotype_evolutionary_distinctiveness | EcoEA | Fitness Sharing | 50 | 50 | 469 | 1.00e-07 | 7.00e-07 | **** | 289111.7 | EcoEA , Fitness Sharing | 1 | 2 | p < 1e-04 |
| mean_phenotype_evolutionary_distinctiveness | EcoEA | Lexicase | 50 | 47 | 0 | 0.00e+00 | 0.00e+00 | **** | 449625.8 | EcoEA , Lexicase | 1 | 3 | p < 1e-04 |
| mean_phenotype_evolutionary_distinctiveness | EcoEA | Random | 50 | 50 | 711 | 2.05e-04 | 2.05e-03 | ** | 610140.0 | EcoEA , Random | 1 | 4 | p = 0.00205 |
| mean_phenotype_evolutionary_distinctiveness | EcoEA | Tournament | 50 | 50 | 569 | 2.70e-06 | 2.72e-05 | **** | 770654.1 | EcoEA , Tournament | 1 | 5 | p < 1e-04 |
| mean_phenotype_evolutionary_distinctiveness | Fitness Sharing | Lexicase | 50 | 47 | 100 | 0.00e+00 | 0.00e+00 | **** | 931168.2 | Fitness Sharing, Lexicase | 2 | 3 | p < 1e-04 |
| mean_phenotype_evolutionary_distinctiveness | Fitness Sharing | Random | 50 | 50 | 2428 | 0.00e+00 | 0.00e+00 | **** | 1091682.4 | Fitness Sharing, Random | 2 | 4 | p < 1e-04 |
| mean_phenotype_evolutionary_distinctiveness | Fitness Sharing | Tournament | 50 | 50 | 1614 | 1.20e-02 | 1.20e-01 | ns | 1252196.5 | Fitness Sharing, Tournament | 2 | 5 | p = 0.12 |
| mean_phenotype_evolutionary_distinctiveness | Lexicase | Random | 47 | 50 | 2350 | 0.00e+00 | 0.00e+00 | **** | 1412710.6 | Lexicase, Random | 3 | 4 | p < 1e-04 |
| mean_phenotype_evolutionary_distinctiveness | Lexicase | Tournament | 47 | 50 | 2255 | 0.00e+00 | 0.00e+00 | **** | 1573224.7 | Lexicase , Tournament | 3 | 5 | p < 1e-04 |
| mean_phenotype_evolutionary_distinctiveness | Random | Tournament | 50 | 50 | 173 | 0.00e+00 | 0.00e+00 | **** | 1733738.9 | Random , Tournament | 4 | 5 | p < 1e-04 |
Looks like everything except fitness sharing and tournament are significantly different from each other.
# Raincloud plot of final mean evolutionary distinctiveness
ggplot(
final_data,
aes(
x=selection_name,
y=mean_phenotype_evolutionary_distinctiveness,
fill=selection_name
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
scale="width"
) +
geom_point(
mapping=aes(color=selection_name),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_log10(
name="Mean evolutionary distinctiveness"
) +
scale_x_discrete(
name="Selection"
) +
scale_fill_discrete(
name="Selection"
) +
scale_color_discrete(
name="Selection"
) +
theme(legend.position = "none")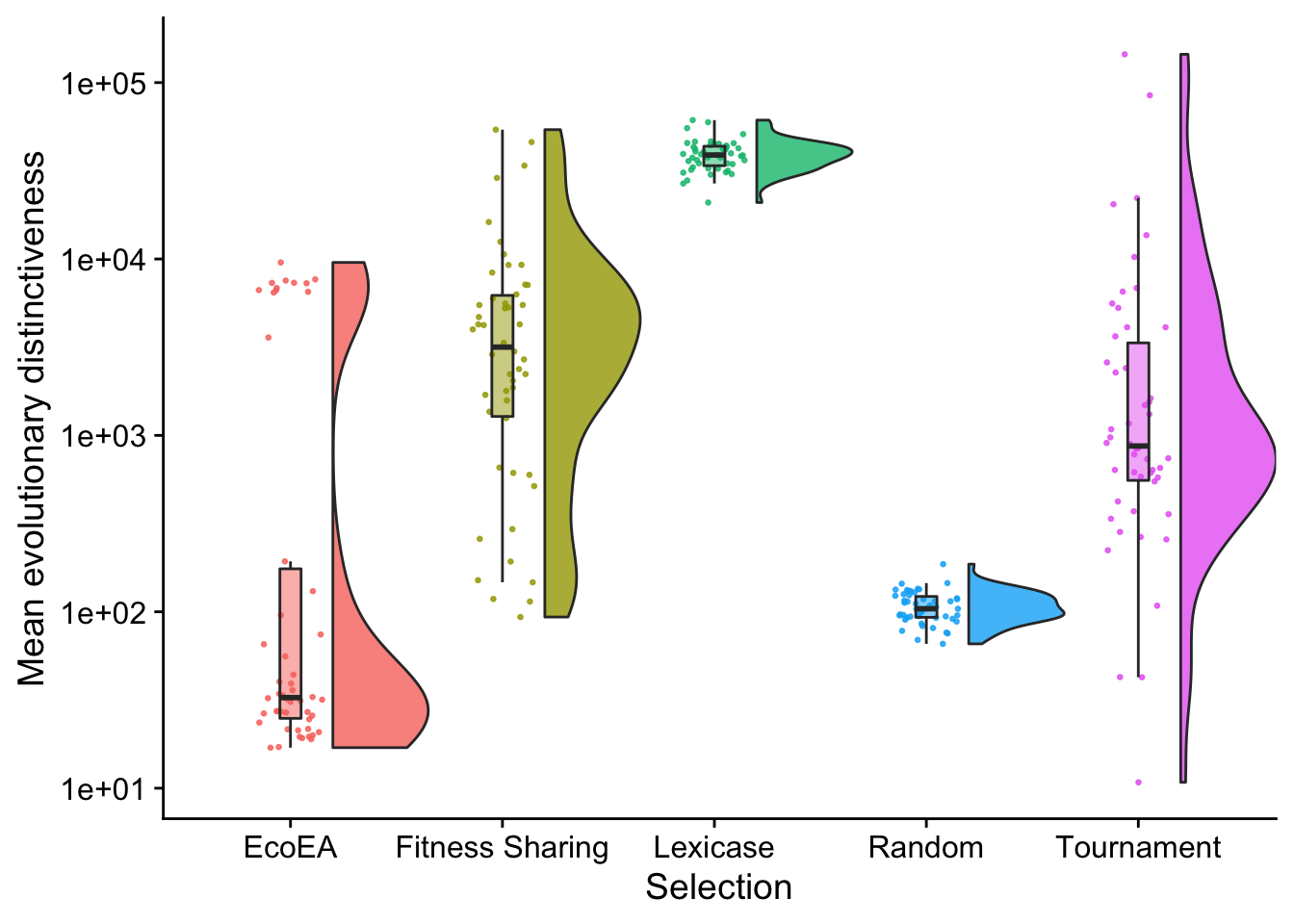
Again, this looks fairly similar to MPD, except that fitness sharing and tournament are higher.
2.4 Phenotypic diversity
Now we analyze phenotypic (i.e. population-level) diversity. Here, we’re defining phenotypes to only include the sites that are actively contributing to fitness. So the phenotype of [1,4,2,6,5,4,3,6,7] would be [0,0,0,6,5,4,3,0,0]. Note that phylogenetic trees are also built using this conception of phenotypes.
2.4.1 Relationship between different types of phenotypic diversity
First, we should assess the extent to which different metrics of phenotypic diversity are capturing different information.
ggplot(
data %>% filter(gen==500000),
aes(
y=phen_diversity,
x=phen_num_taxa,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Phenotypic shannon diversity"
) +
scale_x_continuous(
name="Phenotypic richness",
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")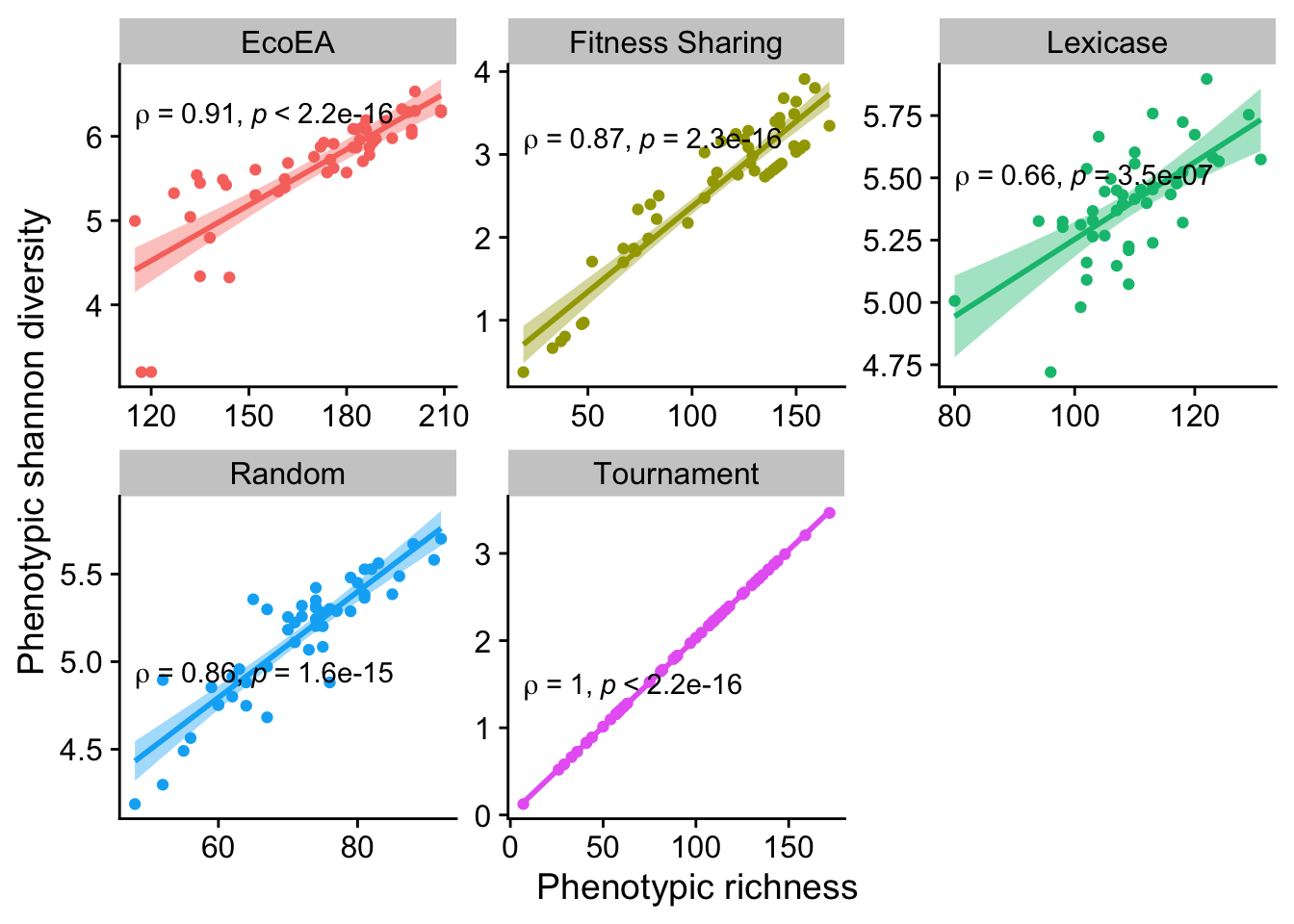
Looks like at the final time point they are pretty much always closely correlated, although this correlation is weaker for lexicase selection than for other selection schemes.
2.4.2 Over time
Now we examine the behavior of each phenotypic diversity metric over time.
2.4.2.1 Richness
ggplot(
data,
aes(
x=gen,
y=phen_num_taxa,
color=selection_name,
fill=selection_name
)
) +
stat_summary(geom="line", fun=mean) +
stat_summary(
geom="ribbon",
fun.data="mean_cl_boot",
fun.args=list(conf.int=0.95),
alpha=0.2,
linetype=0
) +
scale_y_continuous(
name="Phenotypic richness"
) +
scale_x_log10(
name="Generation"
) +
scale_color_discrete("Selection") +
scale_fill_discrete("Selection")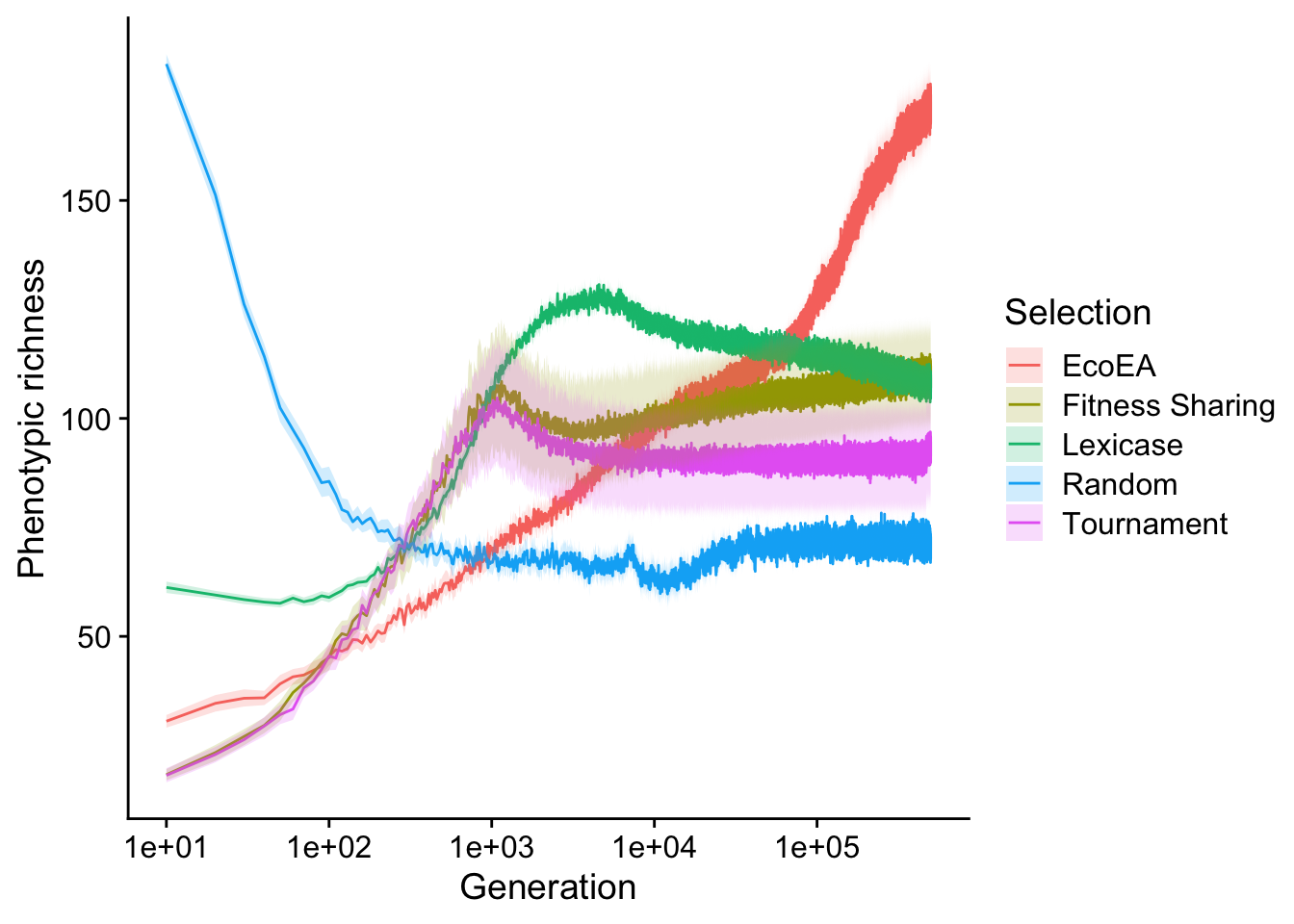
In contrast to the phylodiversity results, phenotypic richness in all selection schemes (even tournament selection) ultimately exceeds that of random selection. Eco-EA monotonically increases while lexicase selection reaches a maximum around the same time it reaches its fitness plateau. The only real similarity to the phylodiversity results is the behavior tournament selection and fitness sharing relative to each other.
2.4.2.2 Shannon diversity
We also looked at phenotypic shannon diversity:
ggplot(
data,
aes(
x=gen,
y=phen_diversity,
color=selection_name,
fill=selection_name
)
) +
stat_summary(geom="line", fun=mean) +
stat_summary(
geom="ribbon",
fun.data="mean_cl_boot",
fun.args=list(conf.int=0.95),
alpha=0.2,
linetype=0
) +
scale_y_continuous(
name="Phenotypic Shannon entropy"
) +
scale_x_log10(
name="Generation"
) +
scale_color_discrete("Selection") +
scale_fill_discrete("Selection")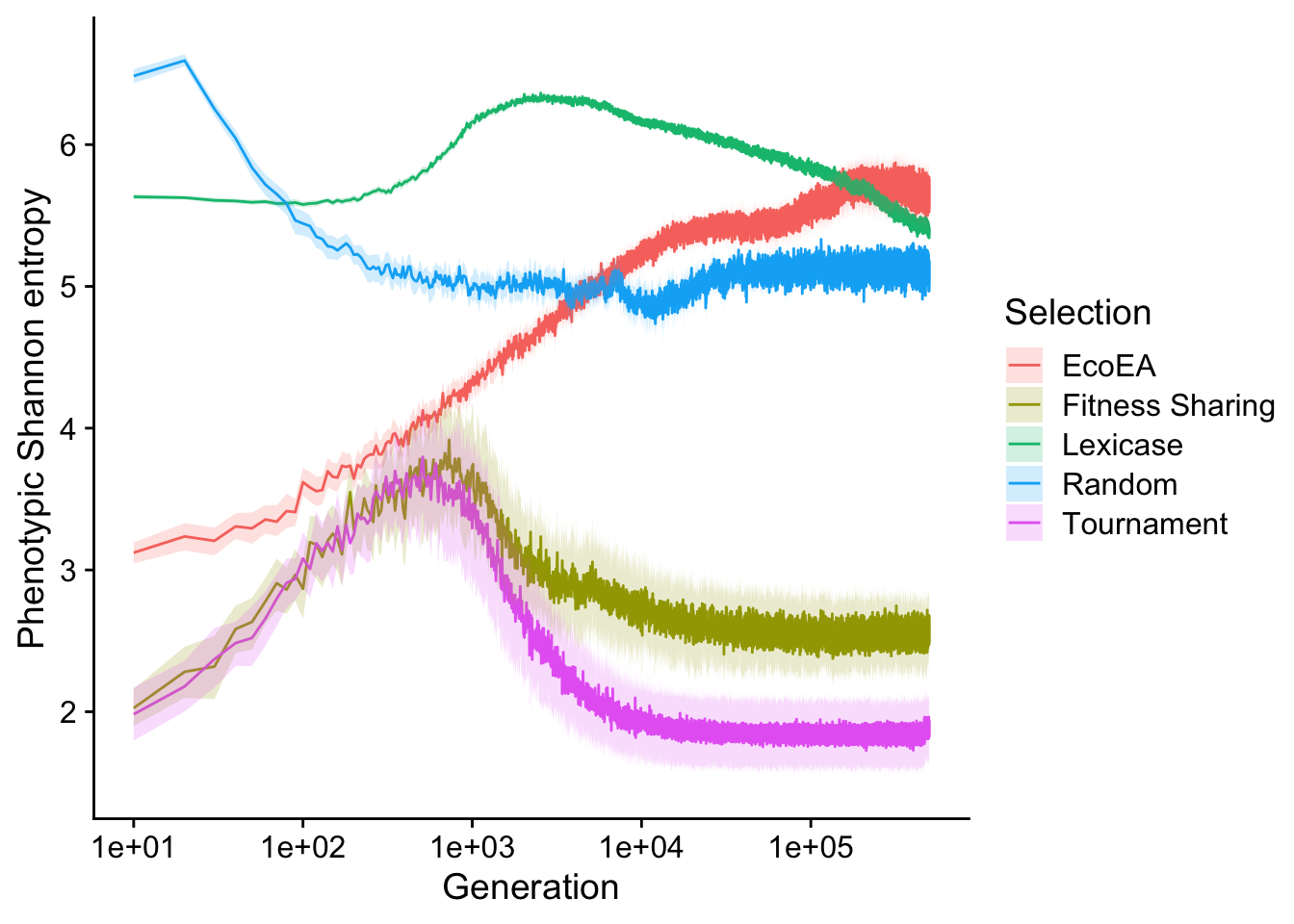
This is more different from the richness results than we might have expected based on the correlation at the final time point. There is a much more pronounced drop off in Shannon entropy for tournament and fitness sharing than in richness. This difference is probably driven by the fact that, after plateauing, these selection schemes are likely both at mutation-selection balance. Thus, there are very many single-mutant phenotypes with only one copy in the population. These do not contribute much to Shannon entropy or phylogenetic diversity, but it does show up in richness.
We also see that, after plataeuing, lexicase selection does actually start to decrease in Shannon entropy, but slowly. Similarly, in Eco-EA, the increase in Shannon entropy towards the end is much more modest than the increase in richness.
2.4.3 Final
Now we assess the final phenotypic diversity
2.4.3.1 Richness
Hypothesis-testing differences between groups:
# Determine which conditions are significantly diferrent from each other
stat.test <- final_data %>%
wilcox_test(phen_num_taxa ~ selection_name) %>%
adjust_pvalue(method = "bonferroni") %>%
add_significance() %>%
add_xy_position(x="selection_name",step.increase=1)
stat.test$label <- mapply(p_label,stat.test$p.adj)
stat.test %>%
kbl() %>%
kable_styling(
bootstrap_options = c(
"striped",
"hover",
"condensed",
"responsive"
)
) %>%
scroll_box(width="600px")| .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | y.position | groups | xmin | xmax | label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| phen_num_taxa | EcoEA | Fitness Sharing | 50 | 50 | 2249.0 | 0.00e+00 | 0.0e+00 | **** | 326 | EcoEA , Fitness Sharing | 1 | 2 | p < 1e-04 |
| phen_num_taxa | EcoEA | Lexicase | 50 | 47 | 2319.0 | 0.00e+00 | 0.0e+00 | **** | 456 | EcoEA , Lexicase | 1 | 3 | p < 1e-04 |
| phen_num_taxa | EcoEA | Random | 50 | 50 | 2500.0 | 0.00e+00 | 0.0e+00 | **** | 586 | EcoEA , Random | 1 | 4 | p < 1e-04 |
| phen_num_taxa | EcoEA | Tournament | 50 | 50 | 2378.5 | 0.00e+00 | 0.0e+00 | **** | 716 | EcoEA , Tournament | 1 | 5 | p < 1e-04 |
| phen_num_taxa | Fitness Sharing | Lexicase | 50 | 47 | 1428.0 | 6.80e-02 | 6.8e-01 | ns | 846 | Fitness Sharing, Lexicase | 2 | 3 | p = 0.68 |
| phen_num_taxa | Fitness Sharing | Random | 50 | 50 | 1973.0 | 6.00e-07 | 6.3e-06 | **** | 976 | Fitness Sharing, Random | 2 | 4 | p < 1e-04 |
| phen_num_taxa | Fitness Sharing | Tournament | 50 | 50 | 1585.0 | 2.10e-02 | 2.1e-01 | ns | 1106 | Fitness Sharing, Tournament | 2 | 5 | p = 0.21 |
| phen_num_taxa | Lexicase | Random | 47 | 50 | 2339.5 | 0.00e+00 | 0.0e+00 | **** | 1236 | Lexicase, Random | 3 | 4 | p < 1e-04 |
| phen_num_taxa | Lexicase | Tournament | 47 | 50 | 1359.0 | 1.85e-01 | 1.0e+00 | ns | 1366 | Lexicase , Tournament | 3 | 5 | p = 1 |
| phen_num_taxa | Random | Tournament | 50 | 50 | 797.0 | 2.00e-03 | 2.0e-02 |
|
1496 | Random , Tournament | 4 | 5 | p = 0.02 |
Raincloud plot:
# Raincloud plot of final phenotypic diversity
final_phenotypic_fig <- ggplot(
final_data,
aes(
x=selection_name,
y=phen_num_taxa,
fill=selection_name
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
scale="width"
) +
geom_point(
mapping=aes(color=selection_name),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
name="Phenotypic Richness"
) +
scale_x_discrete(
name="Selection"
) +
scale_fill_discrete(
name="Selection"
) +
scale_color_discrete(
name="Selection"
) +
theme(legend.position = "none")
final_phenotypic_fig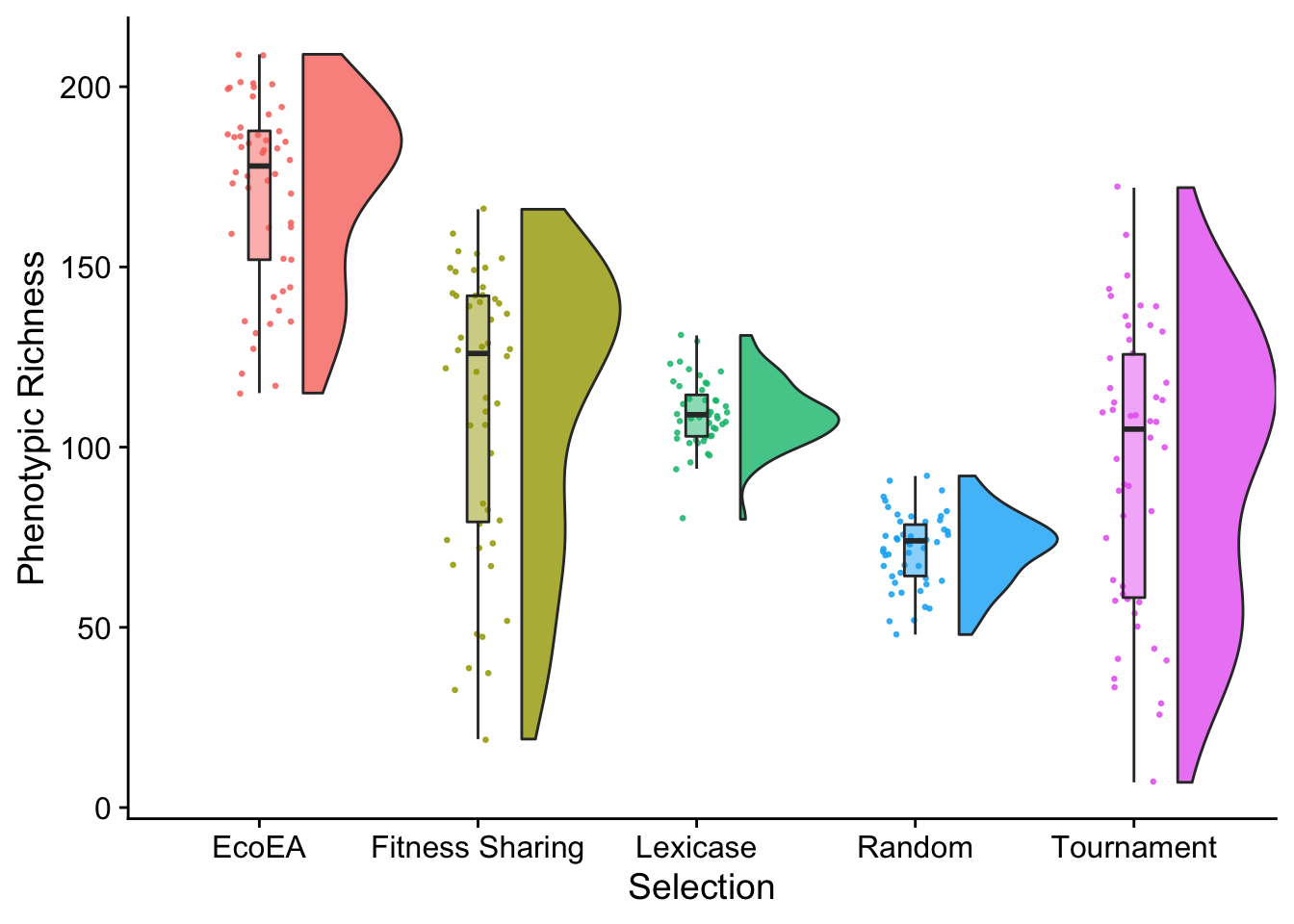
Nothing particularly suprising here, but we should note that, based on the over time plot, this would look a lot different if we had selected a different time point.
2.4.3.2 Shannon diversity
# Determine which conditions are significantly diferrent from each other
stat.test <- final_data %>%
wilcox_test(phen_diversity~ selection_name) %>%
adjust_pvalue(method = "bonferroni") %>%
add_significance() %>%
add_xy_position(x="selection_name",step.increase=1)
stat.test$label <- mapply(p_label,stat.test$p.adj)
stat.test %>%
kbl() %>%
kable_styling(
bootstrap_options = c(
"striped",
"hover",
"condensed",
"responsive"
)
) %>%
scroll_box(width="600px")| .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | y.position | groups | xmin | xmax | label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| phen_diversity | EcoEA | Fitness Sharing | 50 | 50 | 2478.0 | 0.00e+00 | 0.00e+00 | **** | 9.602 | EcoEA , Fitness Sharing | 1 | 2 | p < 1e-04 |
| phen_diversity | EcoEA | Lexicase | 50 | 47 | 1772.0 | 1.66e-05 | 1.66e-04 | *** | 13.012 | EcoEA , Lexicase | 1 | 3 | p = 0.000166 |
| phen_diversity | EcoEA | Random | 50 | 50 | 2089.5 | 0.00e+00 | 1.00e-07 | **** | 16.422 | EcoEA , Random | 1 | 4 | p < 1e-04 |
| phen_diversity | EcoEA | Tournament | 50 | 50 | 2496.0 | 0.00e+00 | 0.00e+00 | **** | 19.832 | EcoEA , Tournament | 1 | 5 | p < 1e-04 |
| phen_diversity | Fitness Sharing | Lexicase | 50 | 47 | 0.0 | 0.00e+00 | 0.00e+00 | **** | 23.242 | Fitness Sharing, Lexicase | 2 | 3 | p < 1e-04 |
| phen_diversity | Fitness Sharing | Random | 50 | 50 | 0.0 | 0.00e+00 | 0.00e+00 | **** | 26.652 | Fitness Sharing, Random | 2 | 4 | p < 1e-04 |
| phen_diversity | Fitness Sharing | Tournament | 50 | 50 | 1856.0 | 2.99e-05 | 2.99e-04 | *** | 30.062 | Fitness Sharing, Tournament | 2 | 5 | p = 0.000299 |
| phen_diversity | Lexicase | Random | 47 | 50 | 1714.0 | 1.01e-04 | 1.01e-03 | ** | 33.472 | Lexicase, Random | 3 | 4 | p = 0.00101 |
| phen_diversity | Lexicase | Tournament | 47 | 50 | 2350.0 | 0.00e+00 | 0.00e+00 | **** | 36.882 | Lexicase , Tournament | 3 | 5 | p < 1e-04 |
| phen_diversity | Random | Tournament | 50 | 50 | 2500.0 | 0.00e+00 | 0.00e+00 | **** | 40.292 | Random , Tournament | 4 | 5 | p < 1e-04 |
Interestingly, the final shannon diversity values of different selection schemes are much more distinguishable from each other than the final richness values.
# Raincloud plot of final phenotypic diversity
ggplot(
final_data,
aes(
x=selection_name,
y=phen_num_taxa,
fill=selection_name
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
scale="width"
) +
geom_point(
mapping=aes(color=selection_name),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
name="Phenotypic Shannon Diversity"
) +
scale_x_discrete(
name="Selection"
) +
scale_fill_discrete(
name="Selection"
) +
scale_color_discrete(
name="Selection"
) +
theme(legend.position = "none")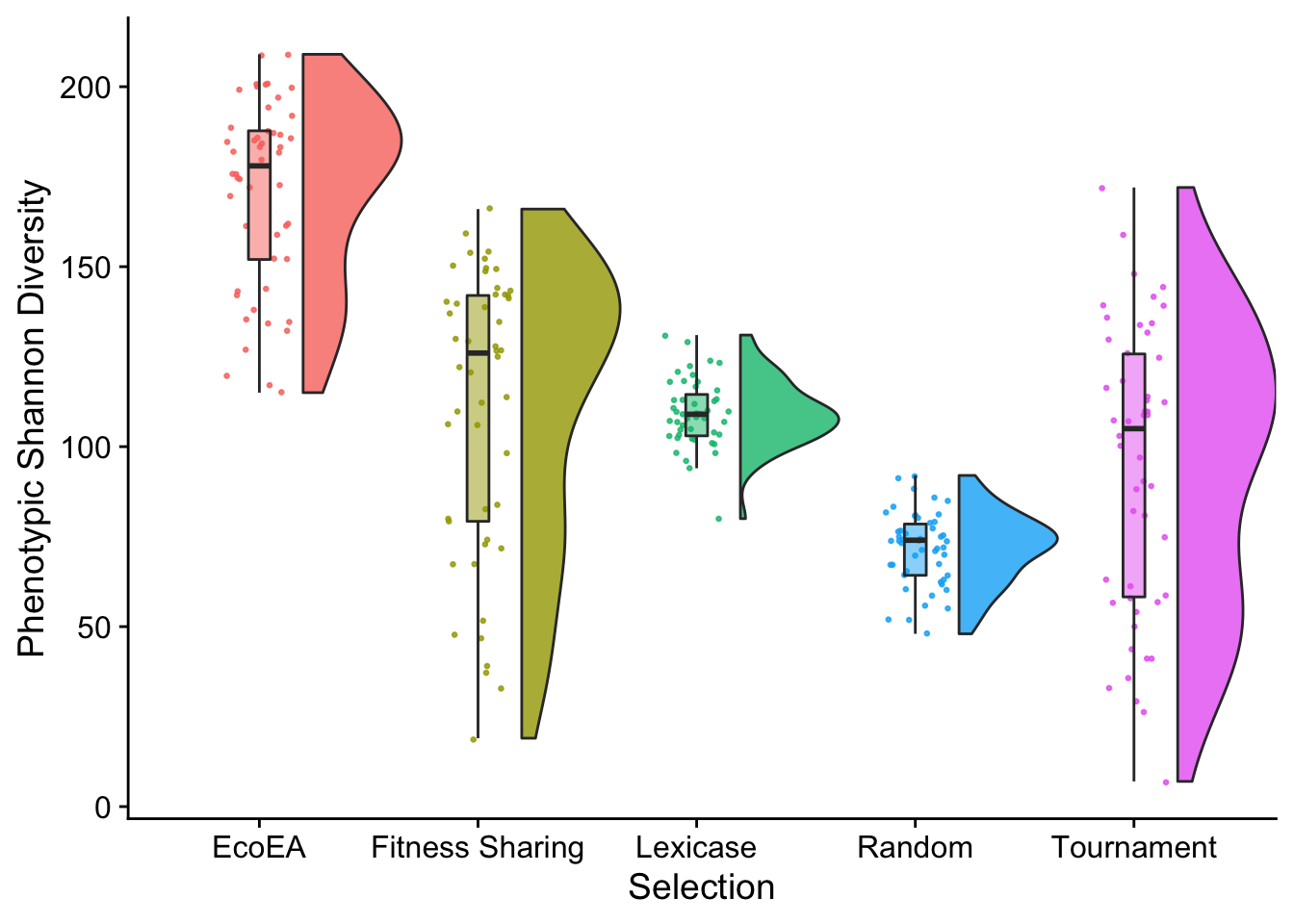
Again, we know from the time series plots that these relative relationships varied a lot over time. Eco-EA is only higher than lexicase selection at the very end.
2.5 Relationship between phenotypic and phylogenetic diversity
Now, we can finally begin to address the main questions. Do phenotypic diversity and phylogenetic diversity capture different information?
2.5.1 Phenotypic richness vs. mean pairwise distance
ggplot(
data %>% filter(gen==500000),
aes(
y=phen_num_taxa,
x=mean_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Phenotypic richness"
) +
scale_x_continuous(
name="Mean pairwise distance",
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")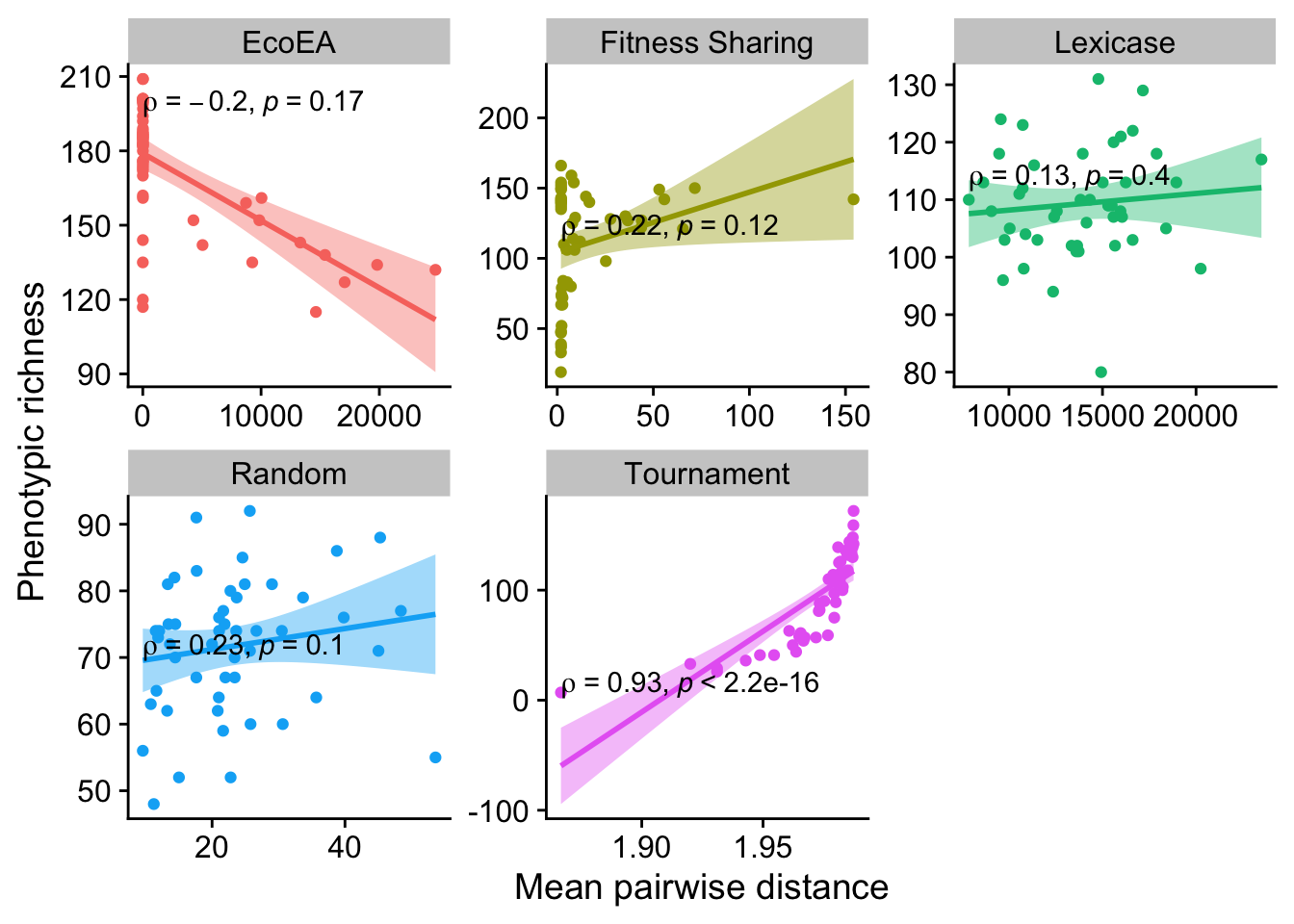
The linear models for some of these are questionable, but the Spearman correlation coefficient should be fine because it does not require linearity. Only tournament selection is significantly different from 0. Eco-EA is even negative (although non-significant), which is probably driven by the fact that there are really two groups of runs in Eco-EA: those that have found a good solution and had their diversity crash, and those that haven’t yet.
Lets take a look at how each of those groups behave (we’ll show fitness as color, just to better understand what’s happening):
ggplot(
data %>% filter(gen==500000, selection_name == "EcoEA"),
aes(
y=phen_num_taxa,
x=mean_phenotype_pairwise_distance,
color = elite_trait_avg
)
) +
geom_point() +
scale_y_continuous(
name="Phenotypic richness"
) +
scale_x_continuous(
name="Mean pairwise distance",
breaks = breaks_extended(4)
) +
facet_wrap(
~mean_phenotype_pairwise_distance > 100, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
scale_color_continuous(type="viridis")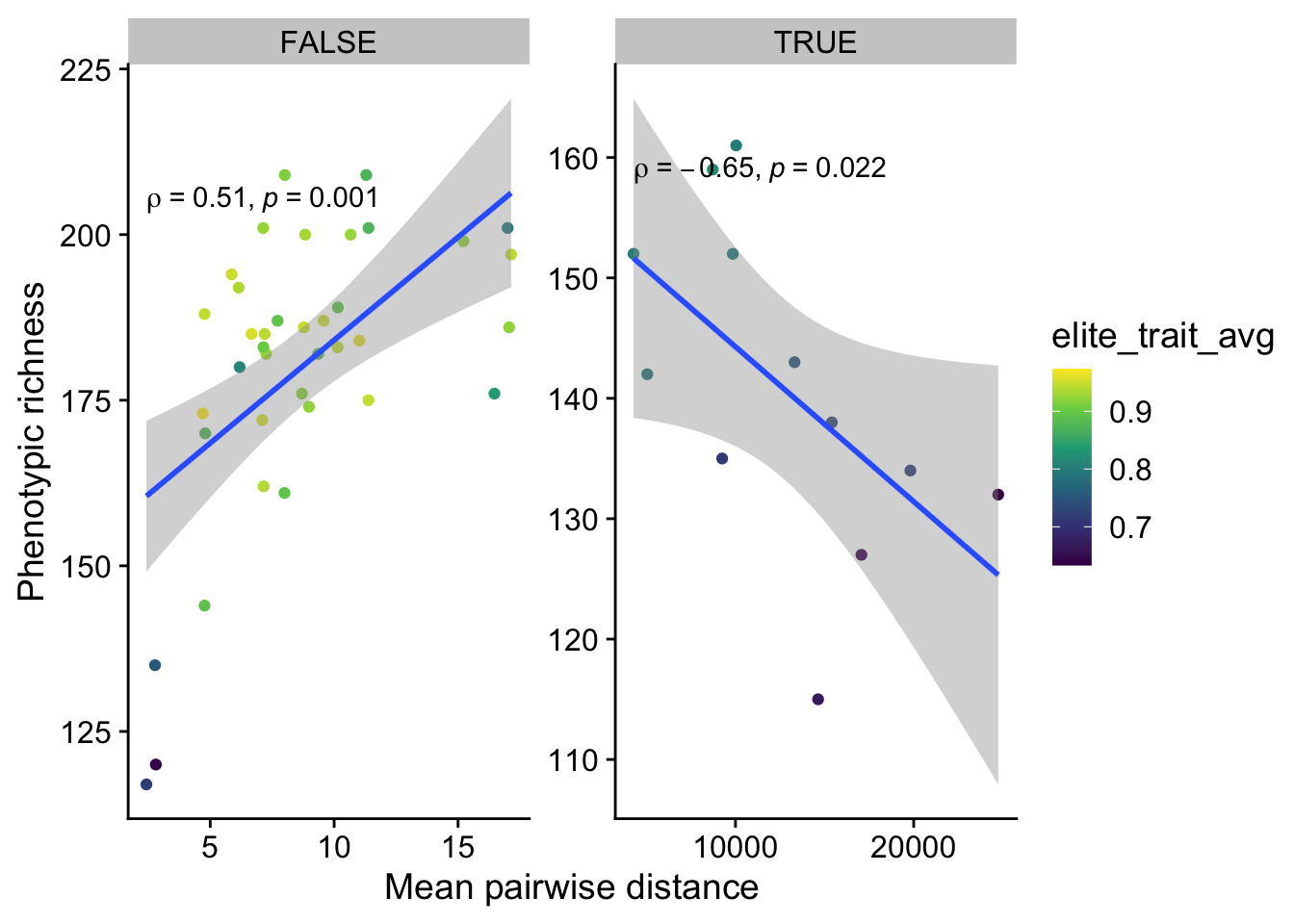
2.5.2 Phenotypic richness vs. mean evolutionary distinctiveness
ggplot(
data %>% filter(gen==500000),
aes(
y=phen_num_taxa,
x=mean_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Phenotypic richness"
) +
scale_x_continuous(
name="Mean evolutionary distinctiveness",
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")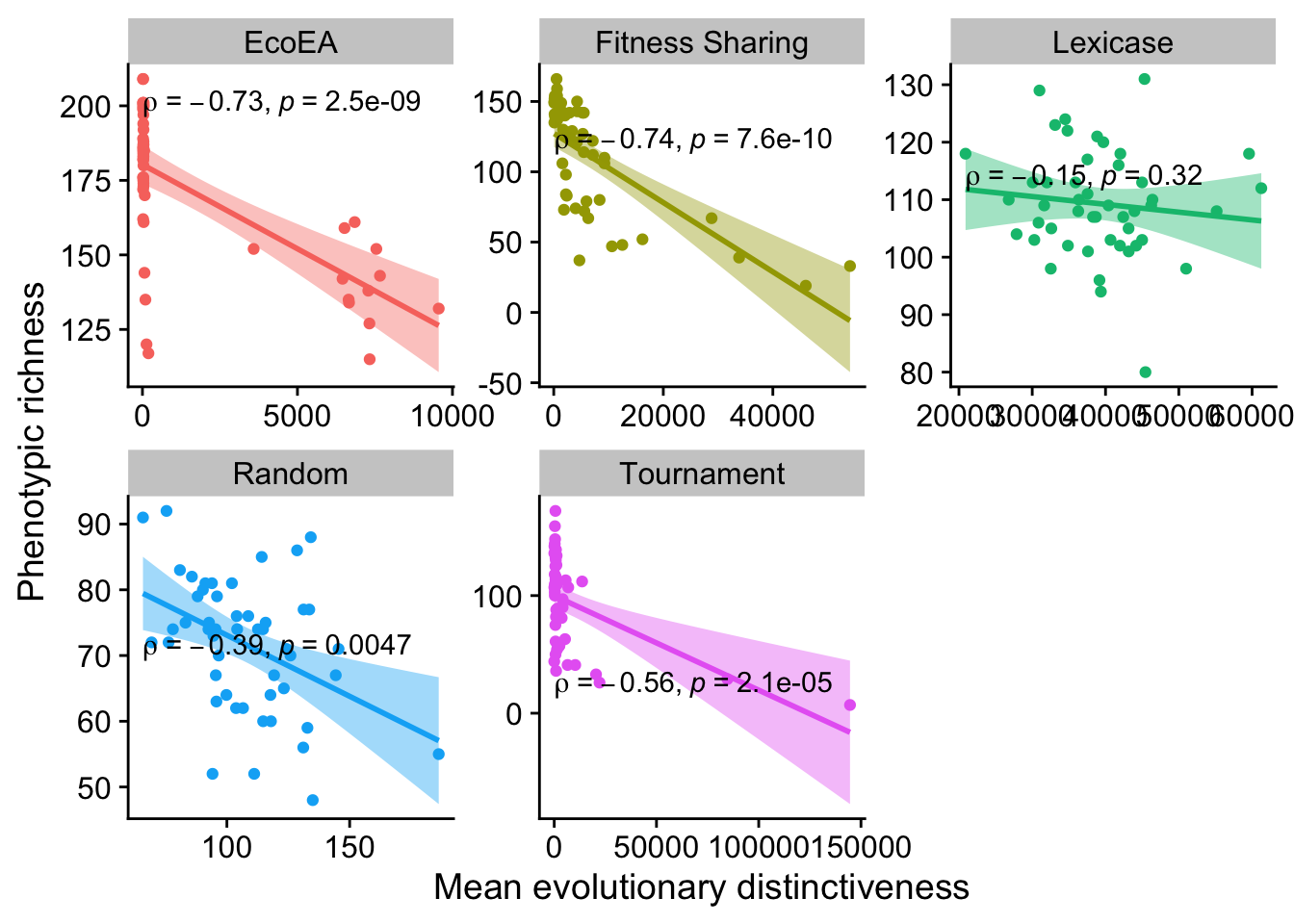
These are mostly significant, but negative. That’s interesting and worthy of further exploration (but it’s a little beyond the scope of this analysis).
2.5.3 Phenotypic shannon diversity vs. mean pairwise distance
ggplot(
data %>% filter(gen==500000),
aes(
y=phen_diversity,
x=mean_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Phenotypic shannon diversity"
) +
scale_x_continuous(
name="Mean pairwise distance",
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")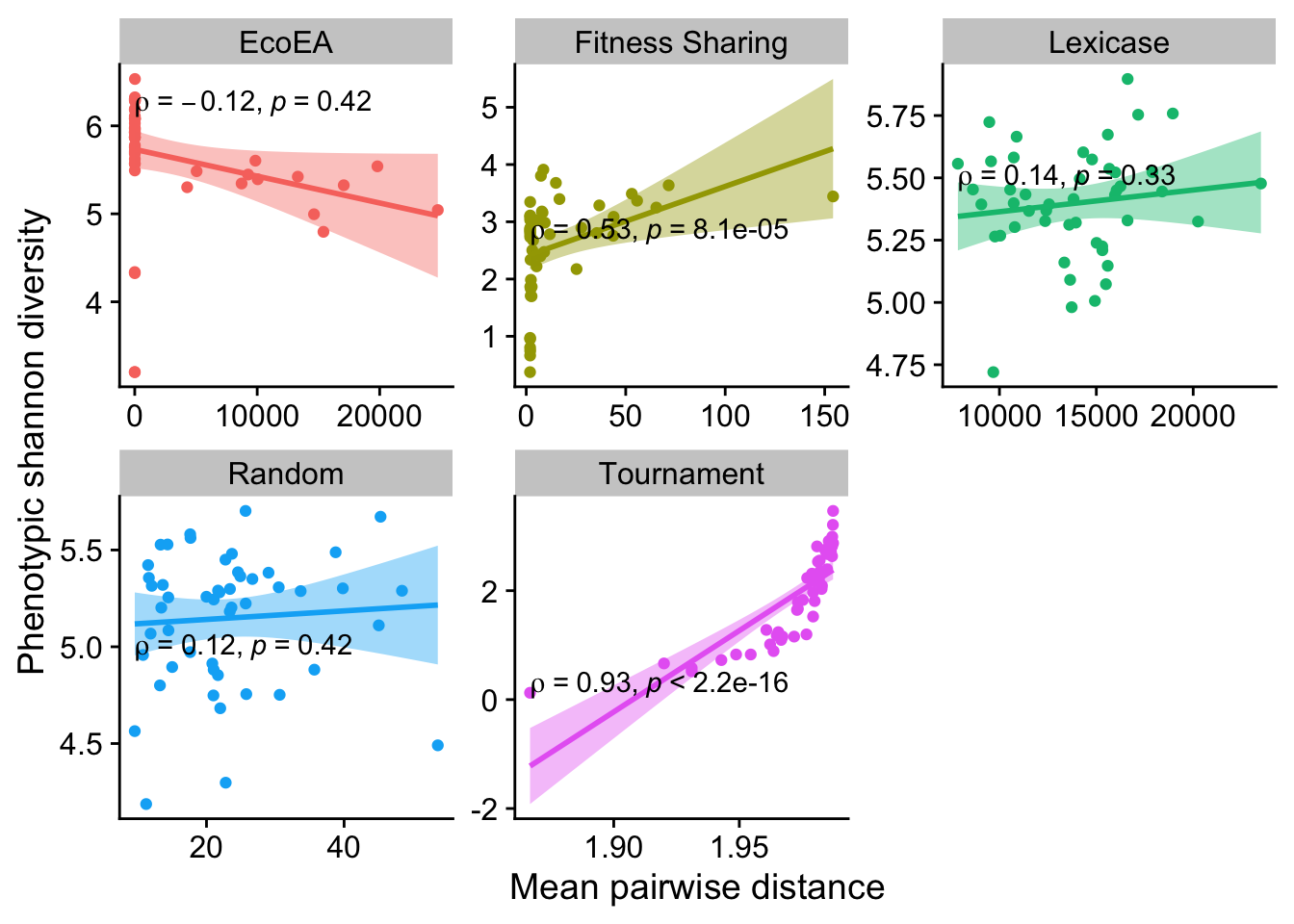
Again, these are mostly not significant (although fitness sharing notably now is).
2.5.4 Phenotypic shannon diversity vs. mean evolutionary distinctiveness
ggplot(
data %>% filter(gen==500000),
aes(
y=phen_diversity,
x=mean_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Phenotypic shannon diversity"
) +
scale_x_continuous(
name="Mean evolutionary distinctiveness",
breaks = breaks_extended(4)
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")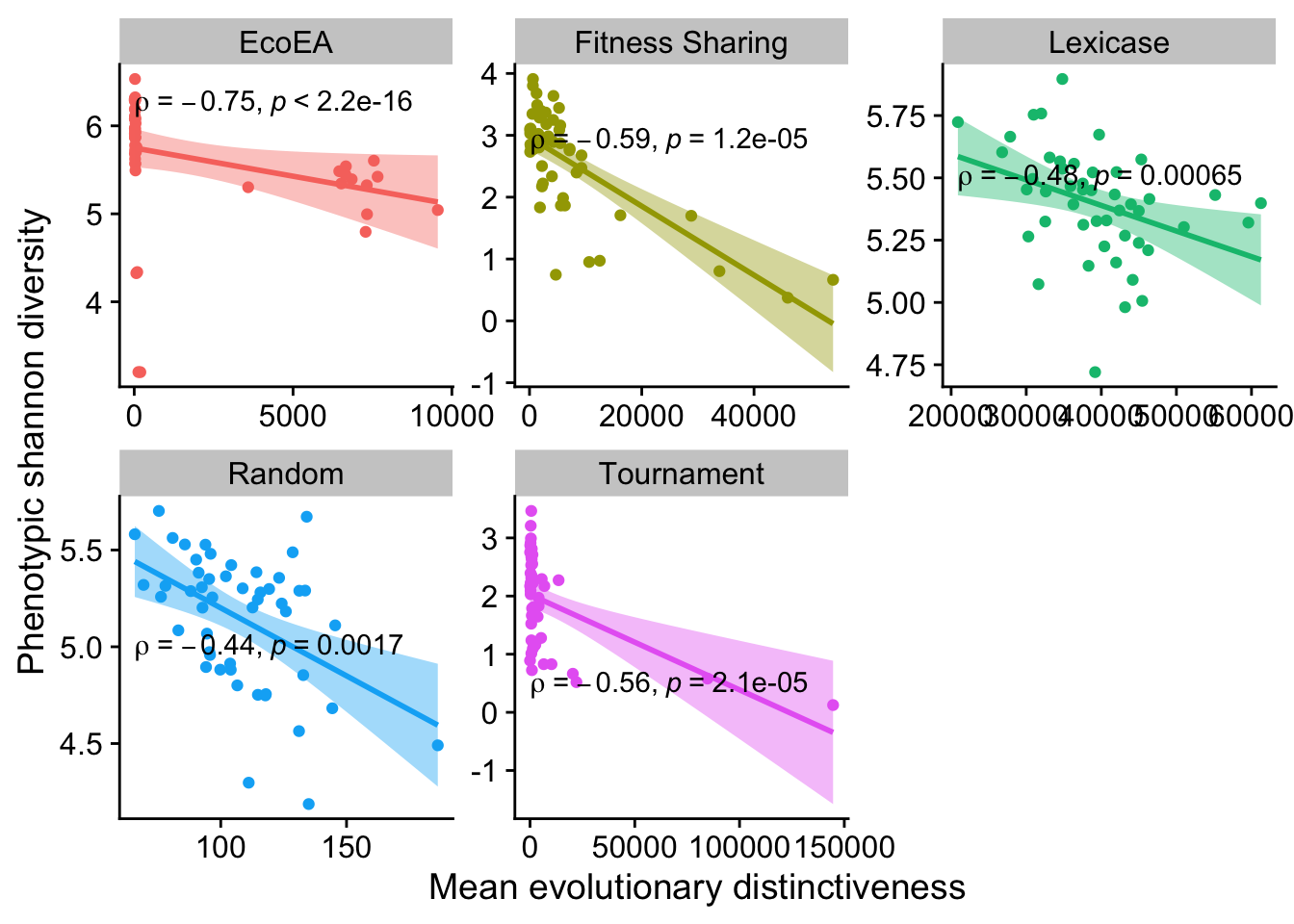
Again, all significant but negative. From this we can conclude that there is substantially more overlap between the information captured by phenotypic diversity and mean evolutionary disinctiveness than between phenotypic diversity metrics and mean pairwise distance. However, thus information has wildly different implications in the two contexts. Moreover, the long term trends are still very different. Thus, we still feel confident in saying that these metrics are meaningfully different. More investigation here may be worthwhile in the future.
Overall, based on these end of time analyses and qualitative comparison of the temporal graphs, we conclude that phenotypic and phylogenetic diversity are meaningfully different from each other in the context of evolutionary computation. In retrospect, this finding may seem obvious. However a lot of people tend to assume that these two classes of diversity are closely related.
2.6 Relationship between diversity and success
At last, we can finally assess whether diversity leads to actually solving problems! We start out by looking at correlations between diversity success across a few different time points, although we ultimately conclude that this approach is not particularly informative.
2.6.1 Very early in run
2.6.1.1 Mean pairwise distance
ggplot(
data %>% filter(gen==25000),
aes(
y=elite_trait_avg,
x=mean_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Mean pairwise distance"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")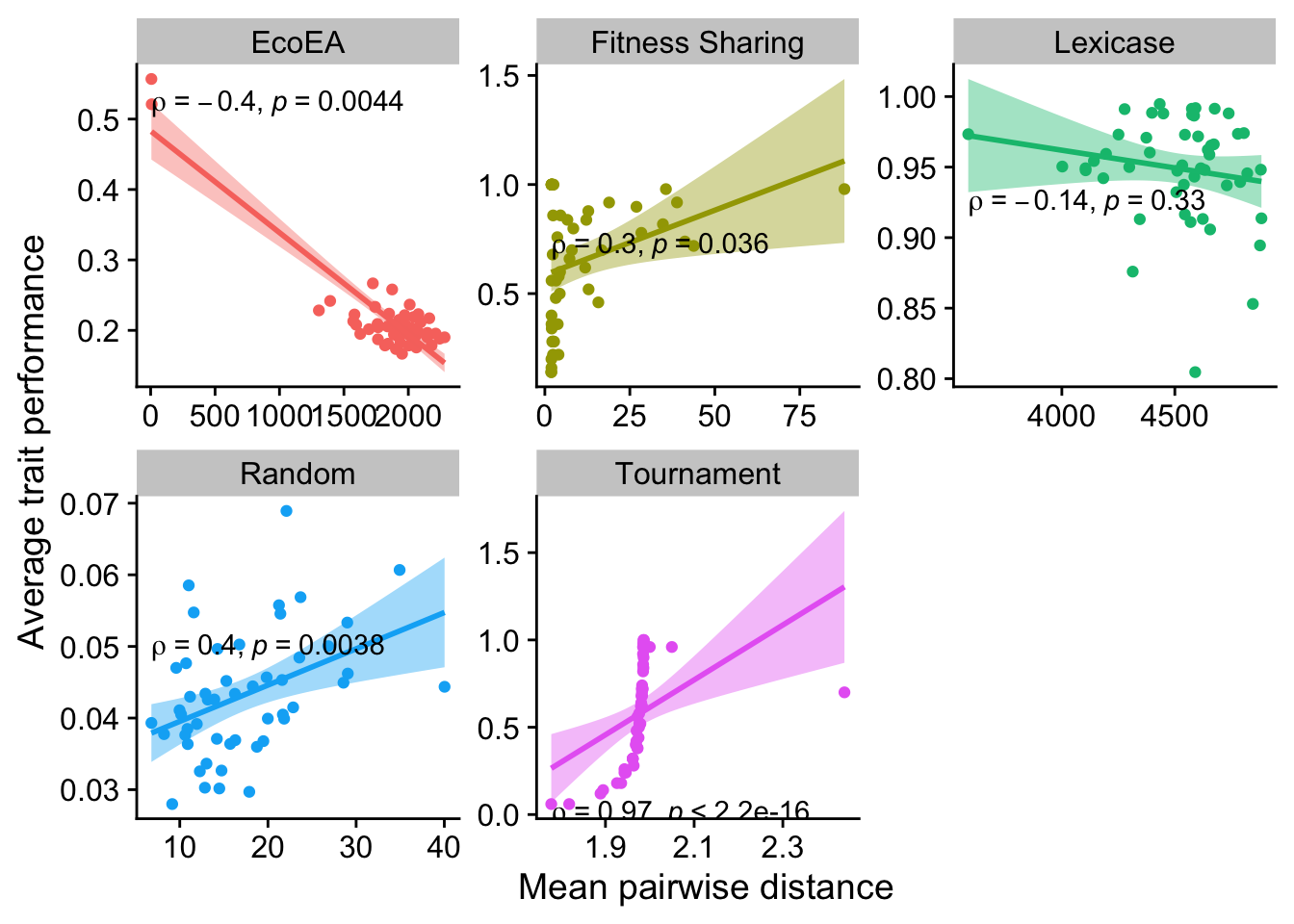
2.6.1.2 Mean evolutionary distinctiveness
ggplot(
data %>% filter(gen==25000),
aes(
y=elite_trait_avg,
x=mean_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Mean evolutionary distinctiveness"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")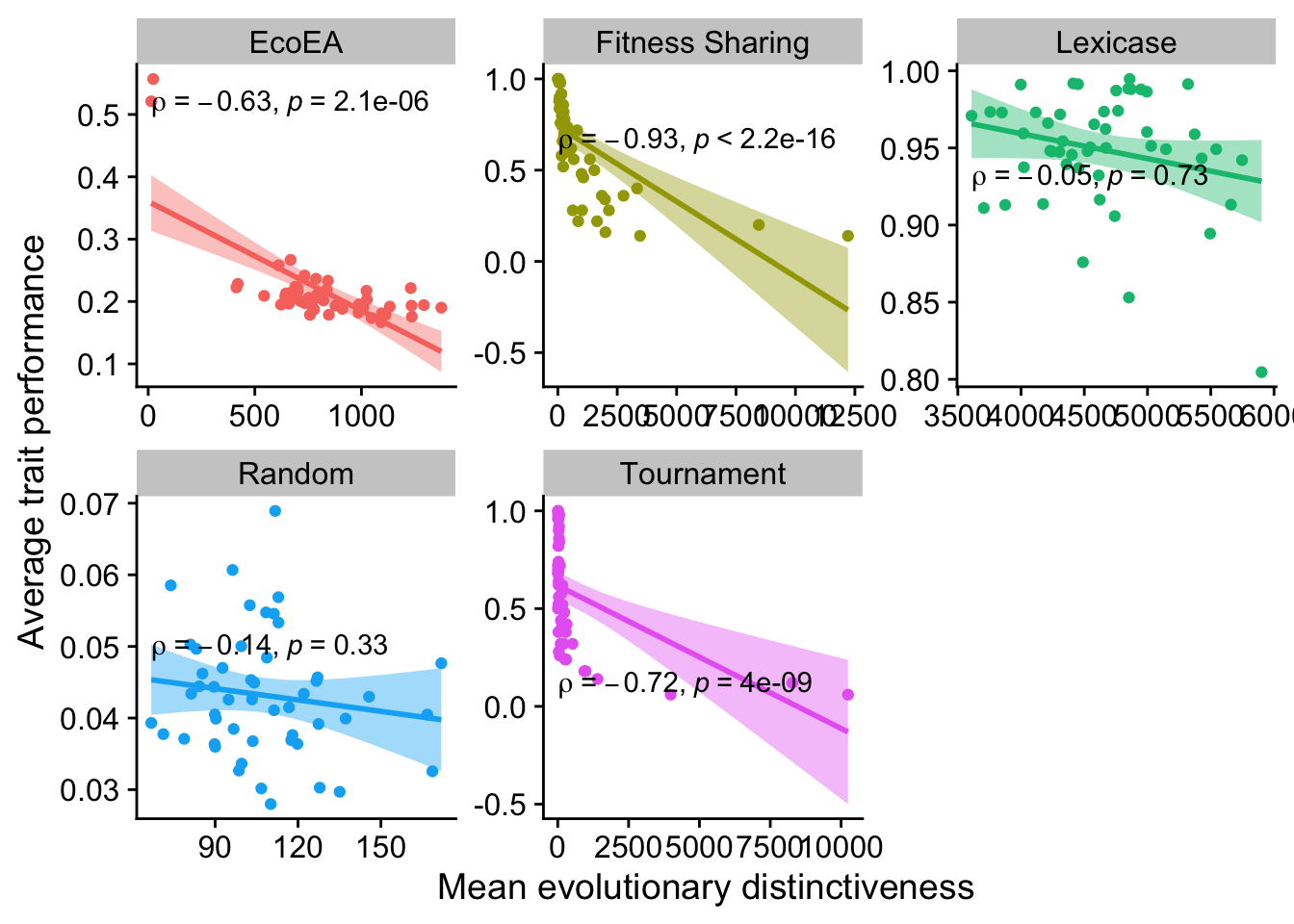
2.6.1.3 Richness
ggplot(
data %>% filter(gen==25000),
aes(
y=elite_trait_avg,
x=phen_num_taxa,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Phenotypic richness"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")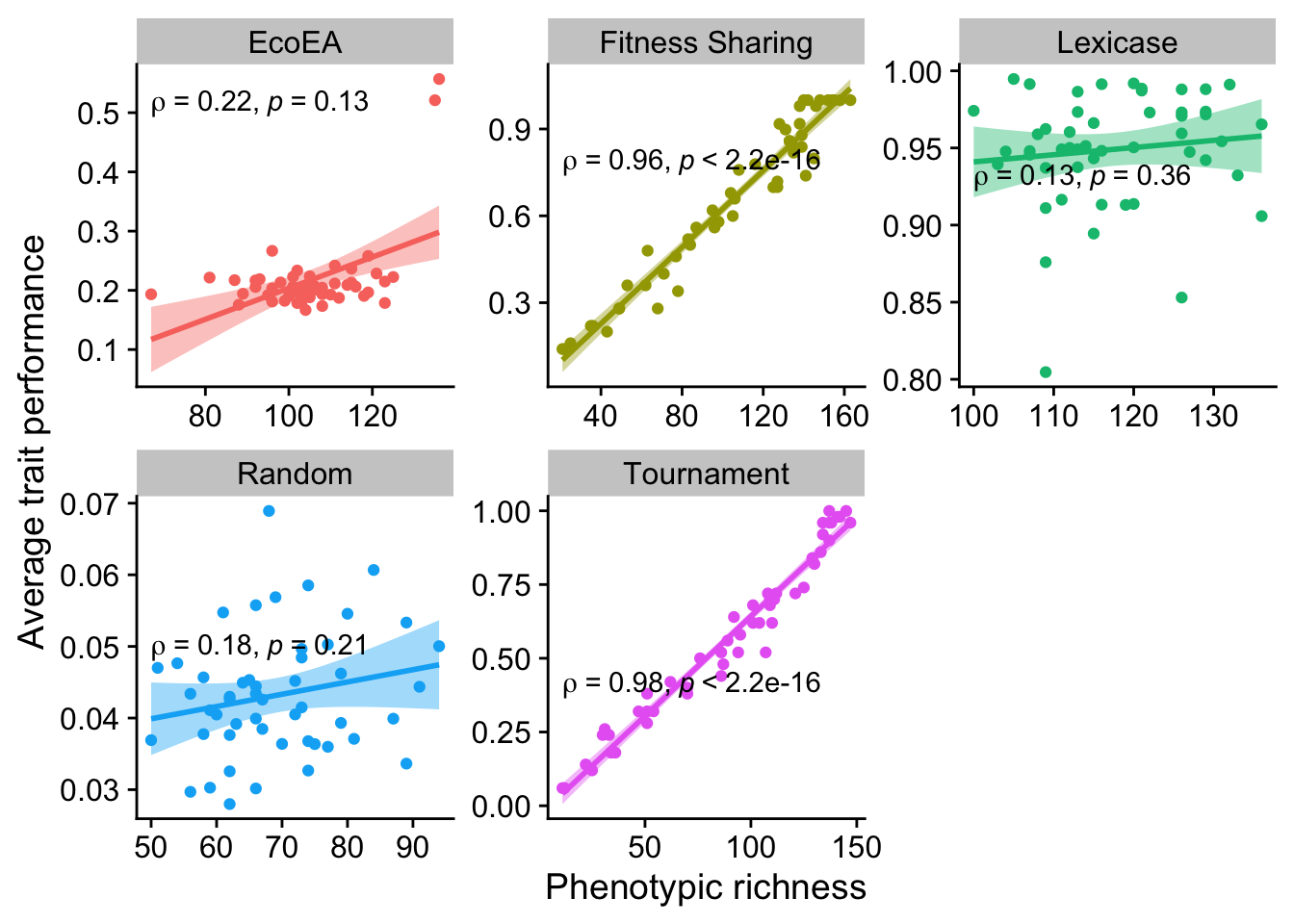
2.6.1.4 Shannon diversity
ggplot(
data %>% filter(gen==25000),
aes(
y=elite_trait_avg,
x=phen_diversity,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Phenotypic Shannon diversity"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")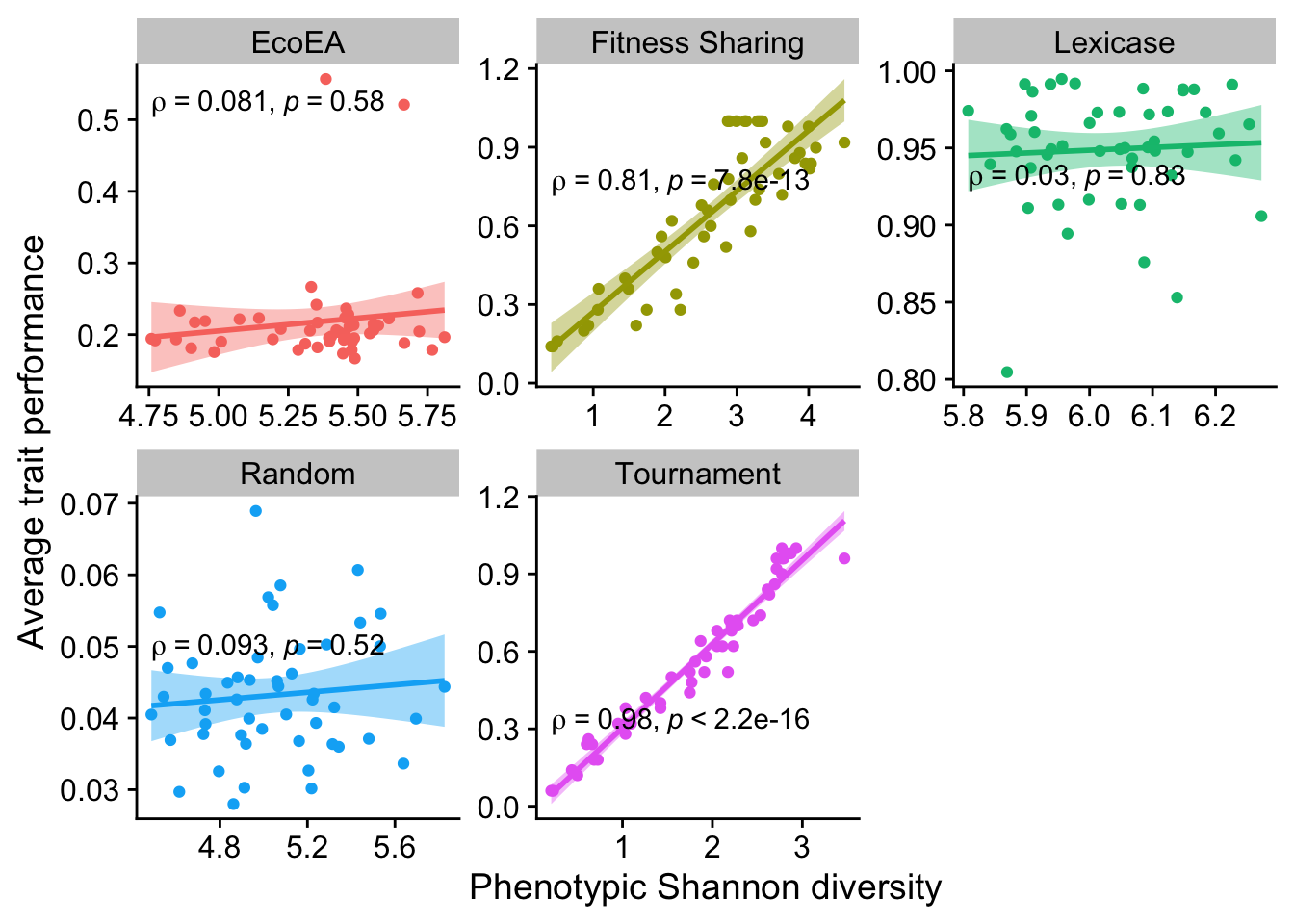
2.6.2 Middle of run
2.6.2.1 Mean pairwise distance
ggplot(
data %>% filter(gen==50000),
aes(
y=elite_trait_avg,
x=mean_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Mean pairwise distance"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")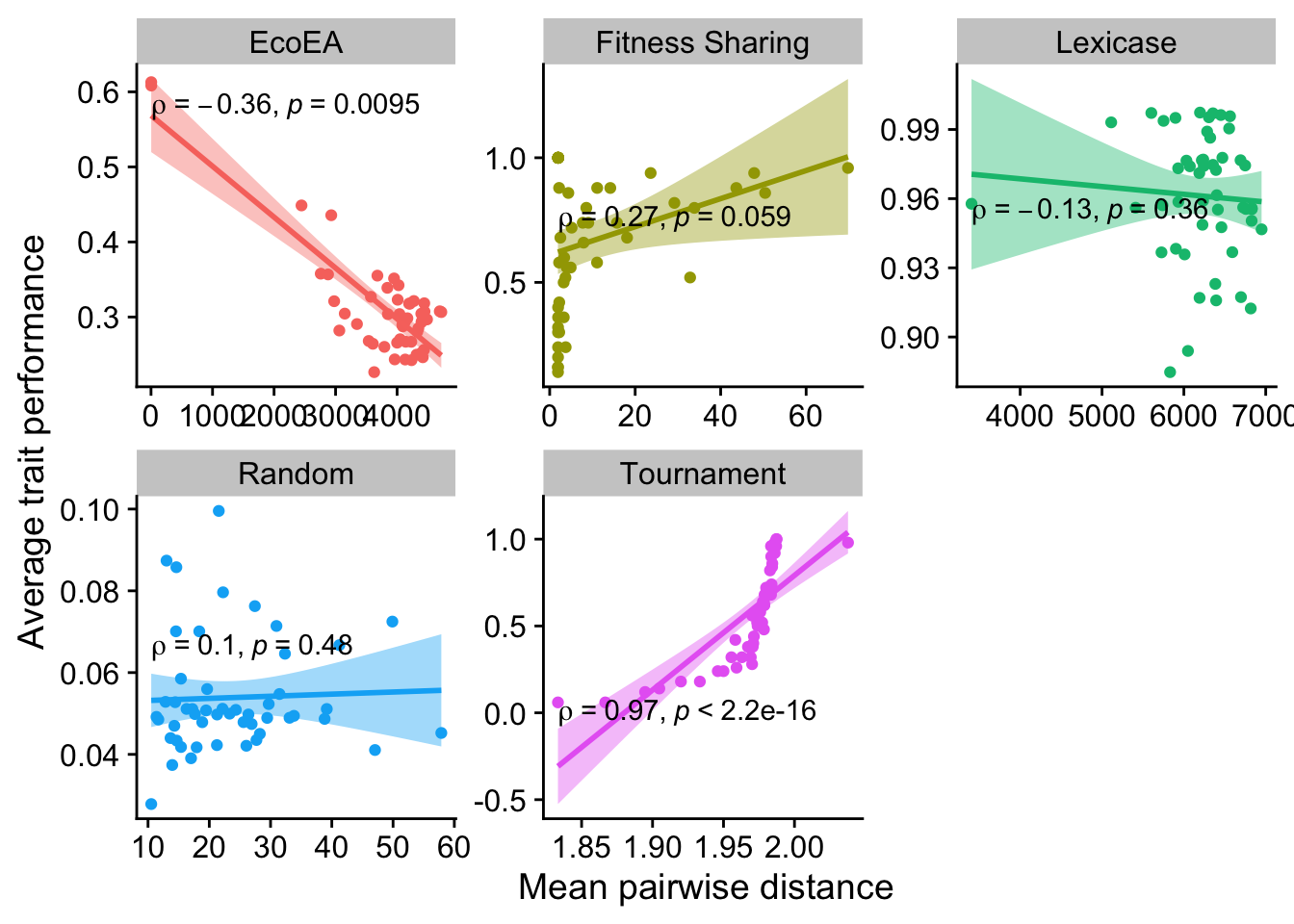
2.6.2.2 Mean evolutionary distinctiveness
ggplot(
data %>% filter(gen==50000),
aes(
y=elite_trait_avg,
x=mean_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Mean evolutionary distinctiveness"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")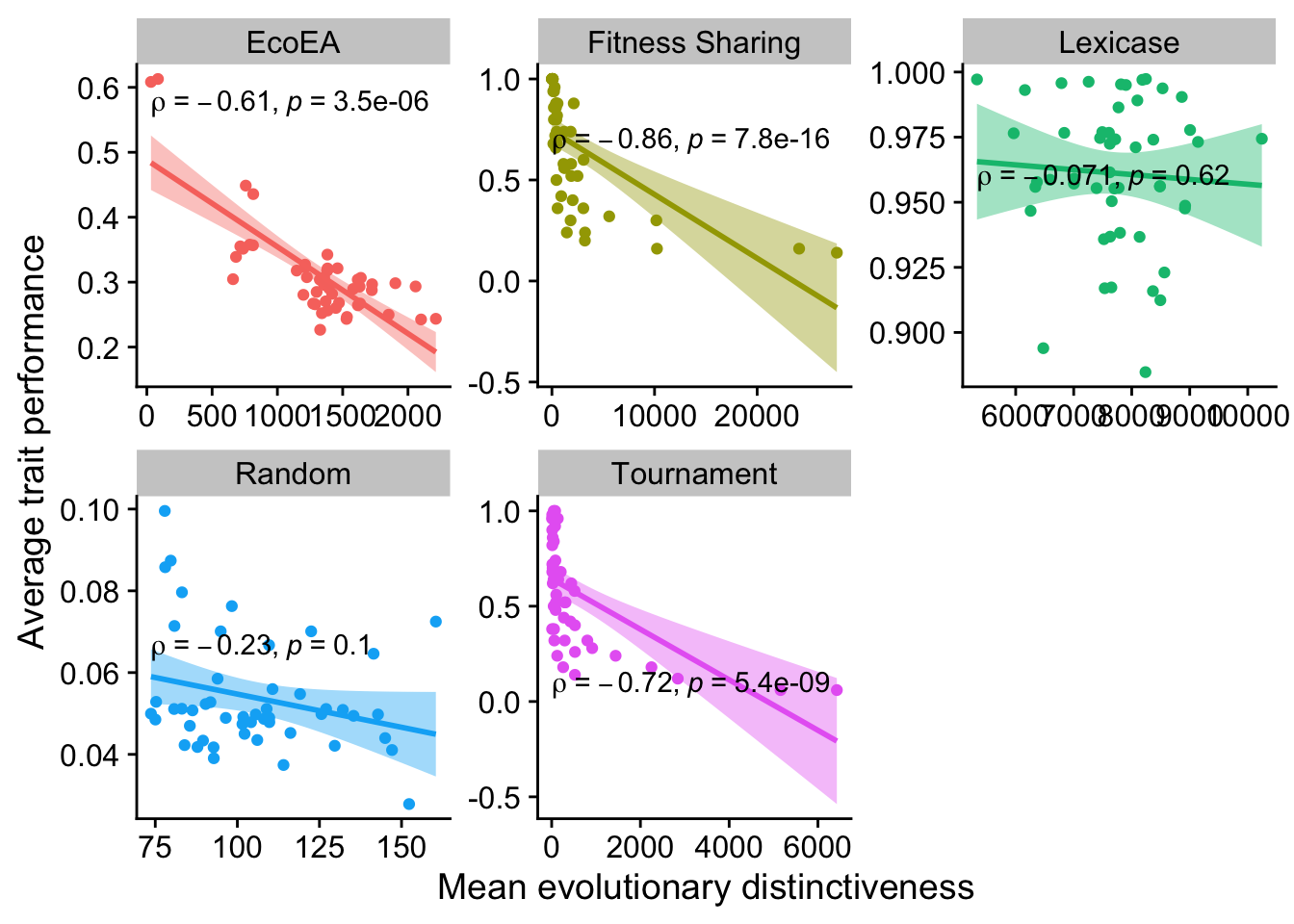
2.6.2.3 Richness
ggplot(
data %>% filter(gen==50000),
aes(
y=elite_trait_avg,
x=phen_num_taxa,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Phenotypic richness"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")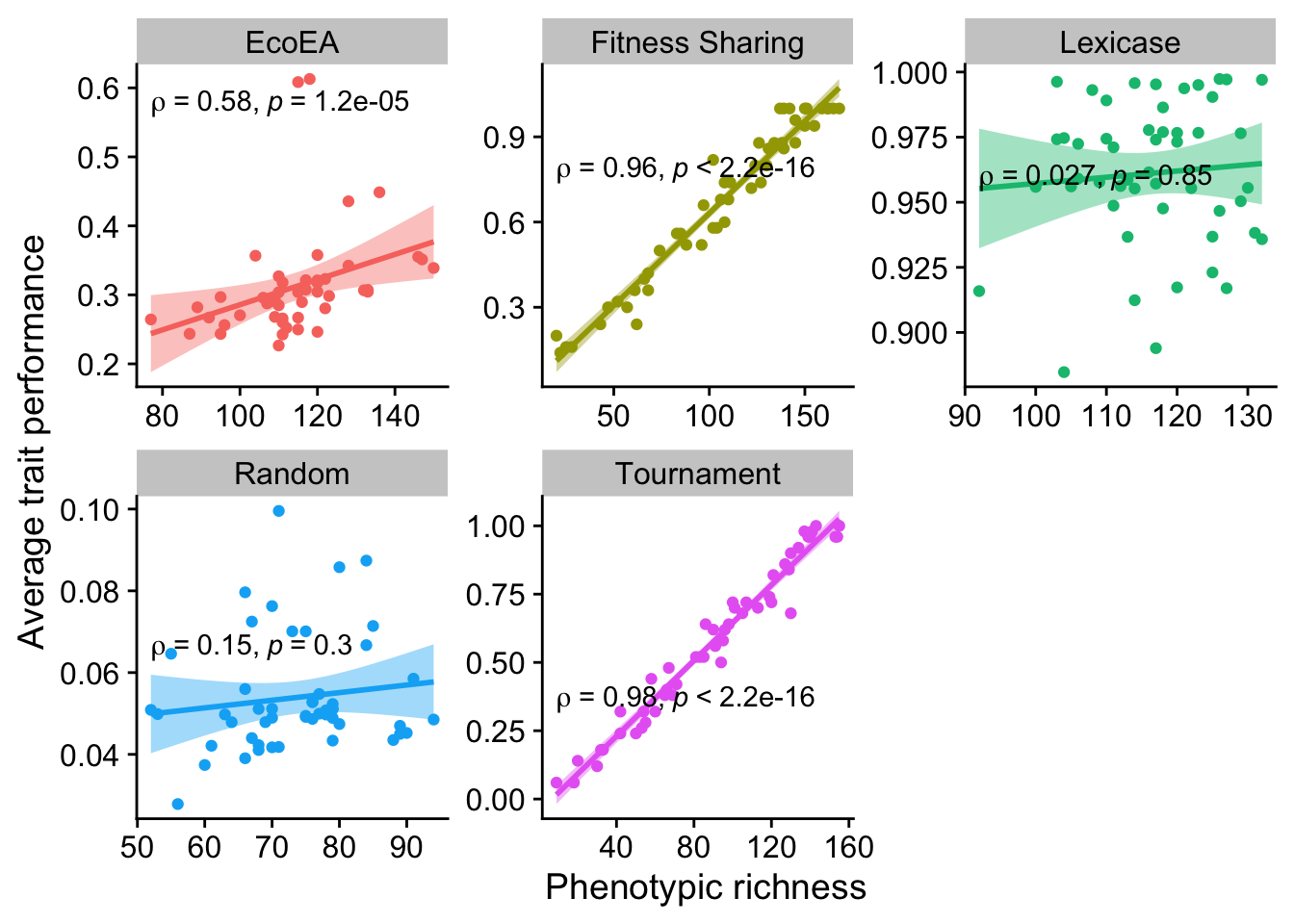
2.6.2.4 Shannon diversity
ggplot(
data %>% filter(gen==50000),
aes(
y=elite_trait_avg,
x=phen_diversity,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Phenotypic Shannon diversity"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")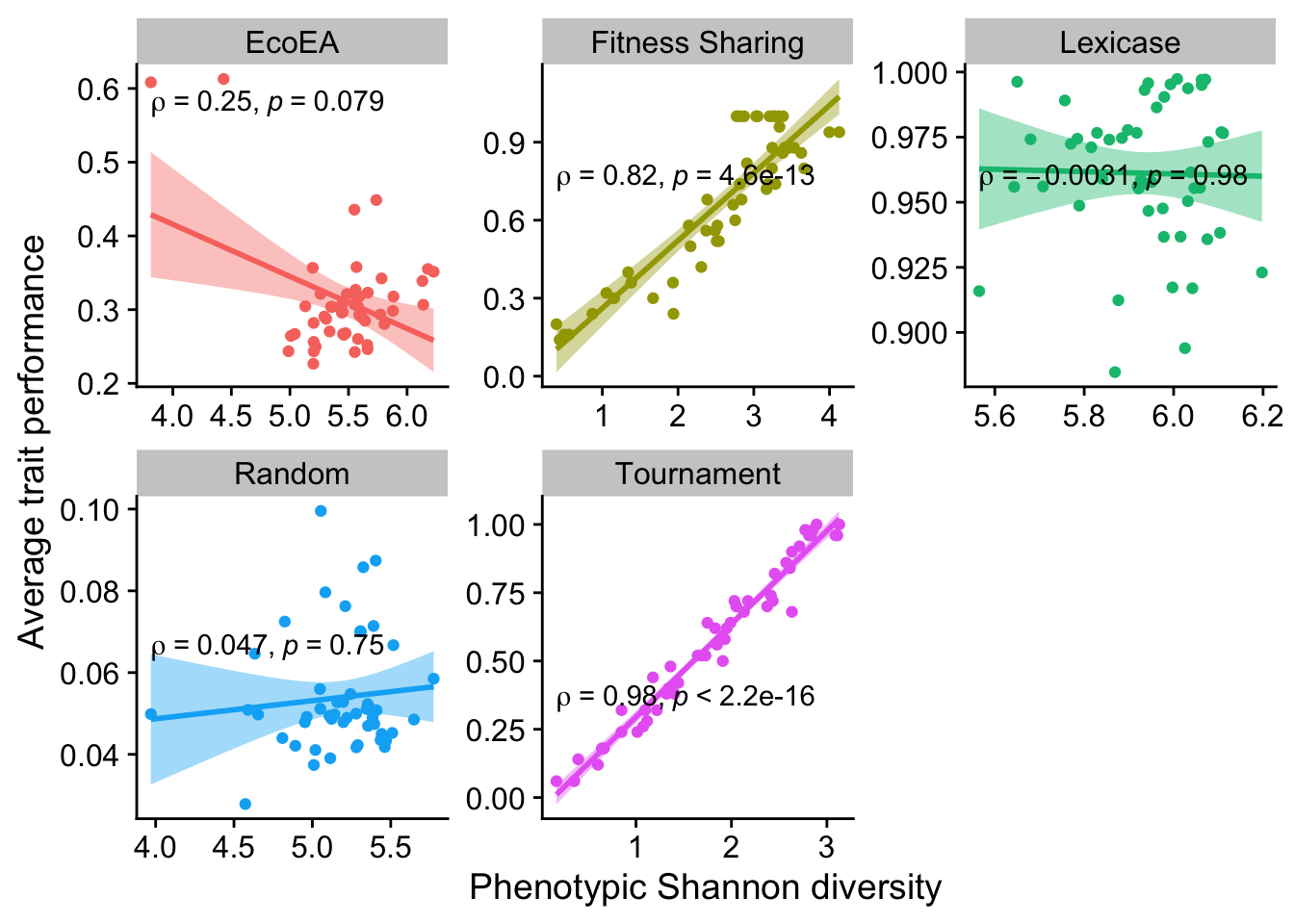
2.6.3 End of run
2.6.3.1 Mean pairwise distance
ggplot(
final_data,
aes(
y=elite_trait_avg,
x=mean_phenotype_pairwise_distance,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Mean pairwise distance"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")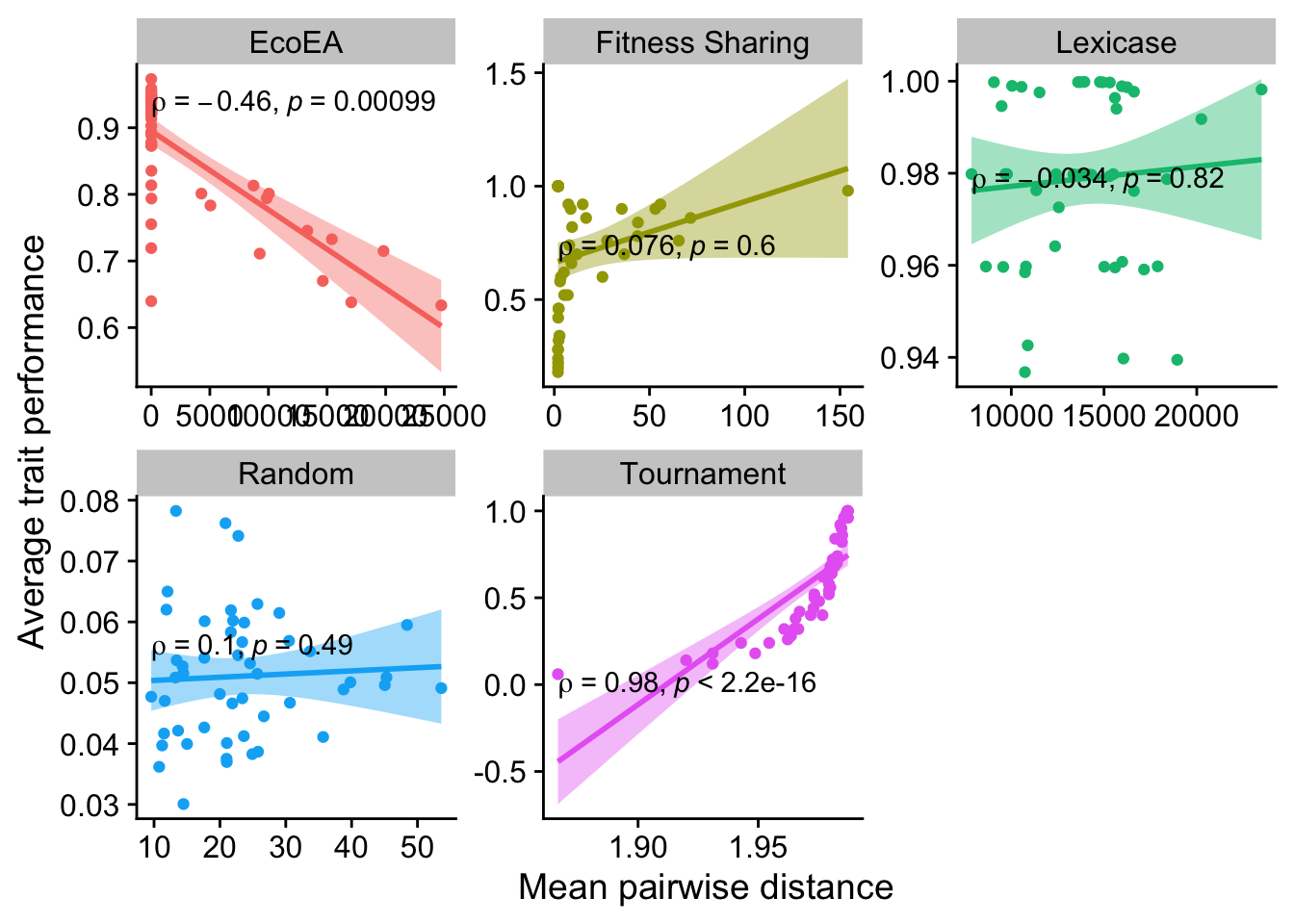
2.6.3.2 Mean evolutionary distinctiveness
ggplot(
final_data,
aes(
y=elite_trait_avg,
x=mean_phenotype_evolutionary_distinctiveness,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Mean evolutionary distinctiveness"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")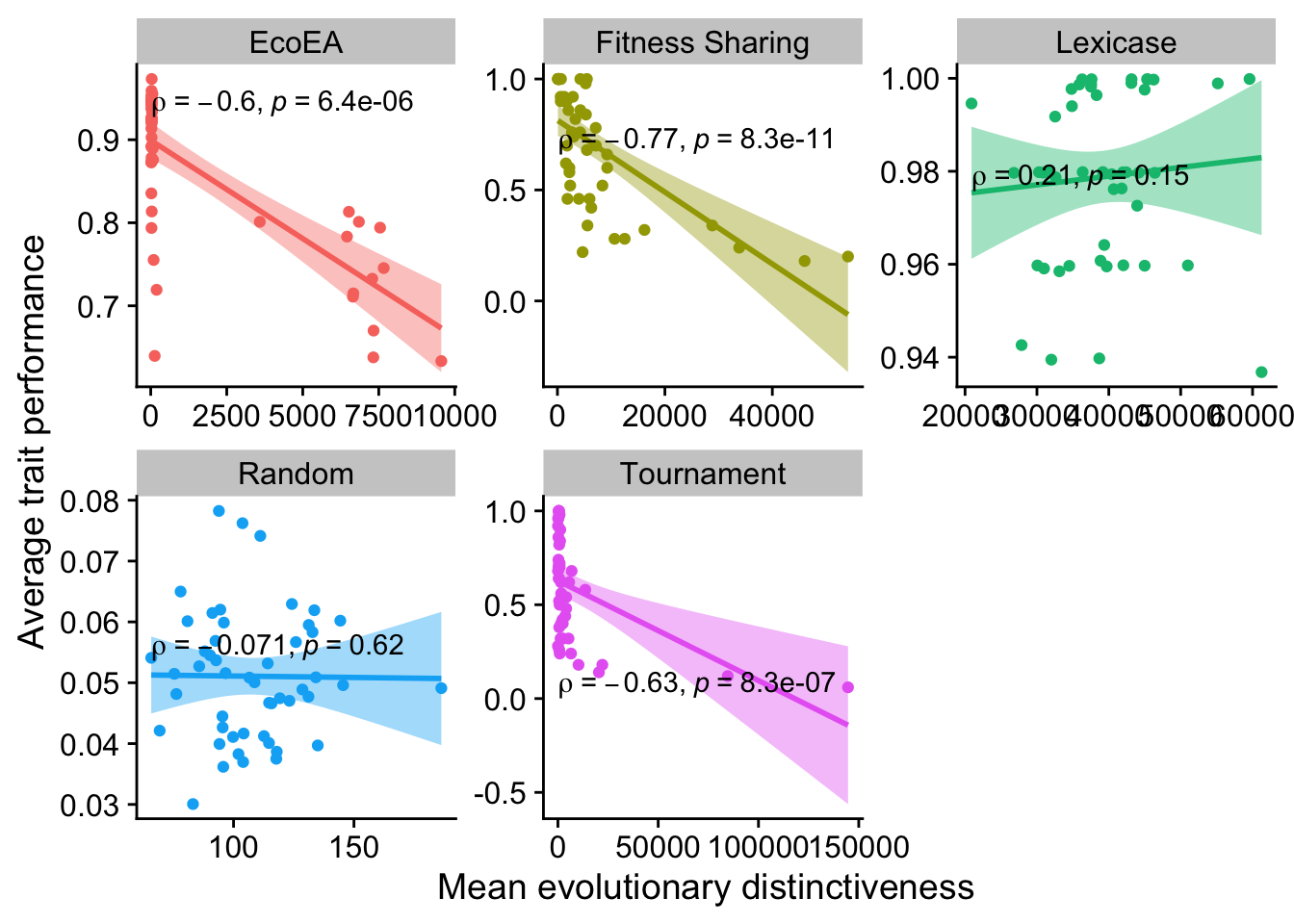
2.6.3.3 Richness
ggplot(
final_data,
aes(
y=elite_trait_avg,
x=phen_num_taxa,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Phenotypic richness"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")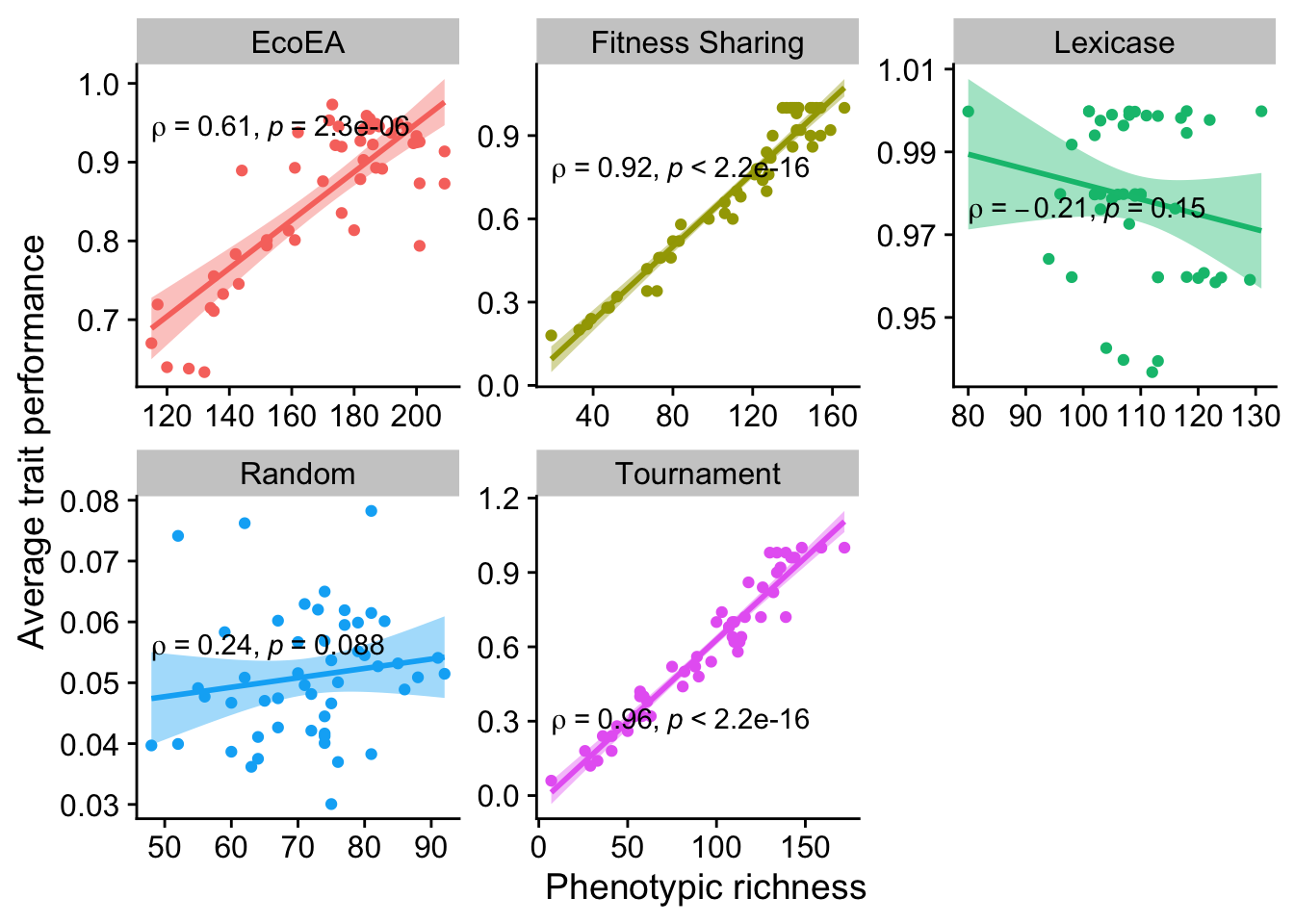
2.6.3.4 Shannon diversity
ggplot(
final_data,
aes(
y=elite_trait_avg,
x=phen_diversity,
color=selection_name,
fill=selection_name
)
) +
geom_point() +
scale_y_continuous(
name="Average trait performance"
) +
scale_x_continuous(
name="Phenotypic Shannon diversity"
) +
facet_wrap(
~selection_name, scales="free"
) +
stat_smooth(
method="lm"
) +
stat_cor(
method="spearman", cor.coef.name = "rho", color="black"
) +
theme(legend.position = "none")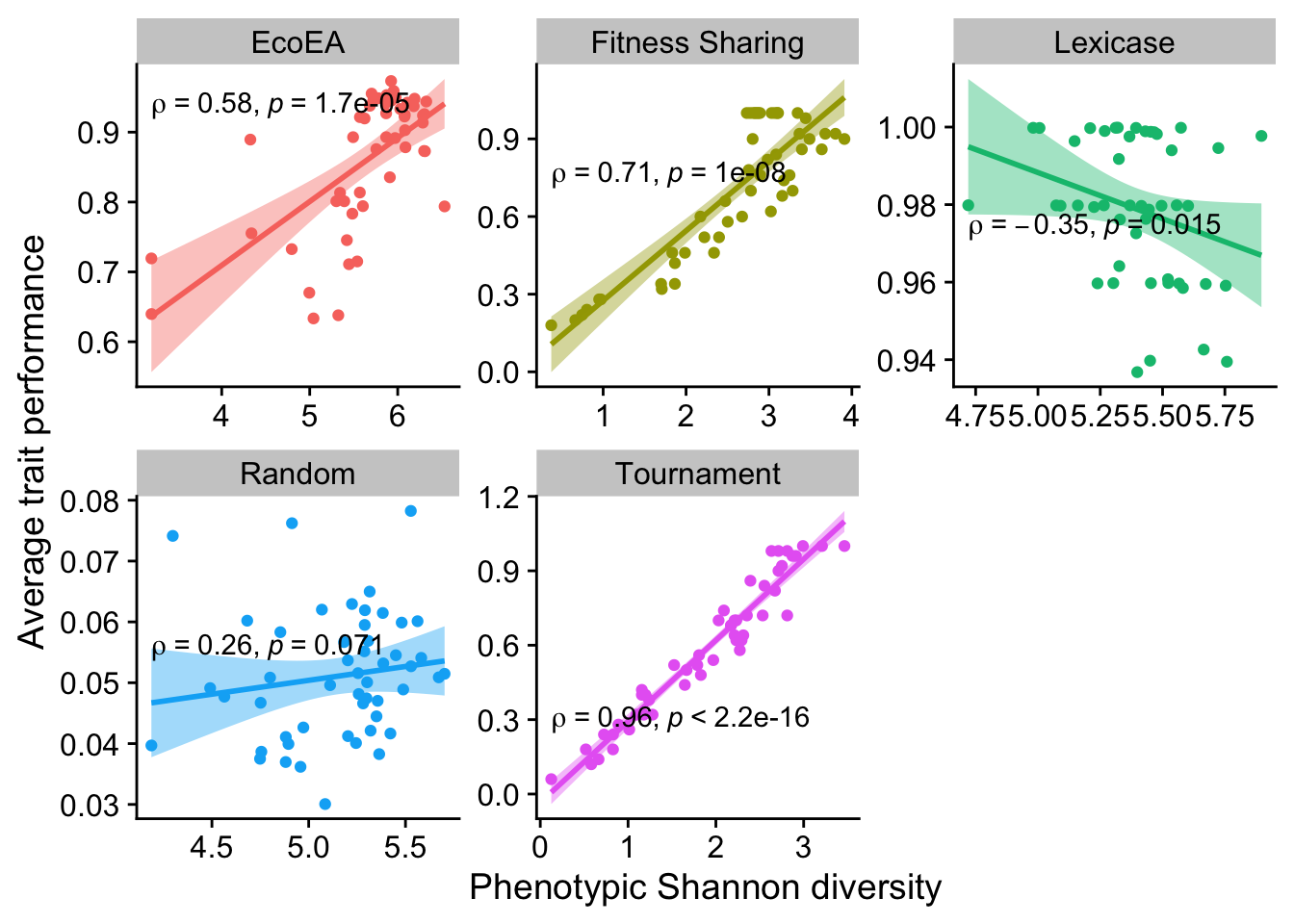
2.7 Causality analysis
Ultimately, it’s hard to draw useful inferences either from these single time point analyses or from comparison of line plots. Due to the feedbacks between diversity and performance, there may be a time delay in when one affects the other.
To analyze the drivers of this feedback loop in a more rigorous way, we turn to Transfer Entropy as a metric of Granger Causality. For a more thorough description, see the paper associated with this supplement.
2.7.1 Setup
First let’s define a function we’ll use to calculate and output significance and effect size for these results:
transfer_entropy_stats <- function(res) {
stat.test <- res %>%
group_by(selection_name, offset) %>%
wilcox_test(value ~ Type) %>%
adjust_pvalue(method = "bonferroni") %>%
add_significance()
stat.test$label <- mapply(p_label,stat.test$p.adj)
# Calculate effect sizes for these differences
effect_sizes <- res %>%
group_by(selection_name, offset) %>%
wilcox_effsize(value ~ Type)
stat.test$effsize <- effect_sizes$effsize
stat.test$magnitude <- effect_sizes$magnitude
stat.test %>%
kbl() %>%
kable_styling(
bootstrap_options = c(
"striped",
"hover",
"condensed",
"responsive"
)
) %>%
scroll_box(width="600px")
}2.7.2 Transfer entropy from diversity to fitness
First, we calculate the information that each type of diversity gives us about future fitness.
2.7.2.1 Max pairwise distance vs. phenotypic richness
We plot the differences in transfer entropy from phylogenetic diversity to fitness and from phenotypic diversity to fitness, at a range of different lags.
# Calculate transfer entropy for max pairwise distance
# Time points are 10 generations, so calculating lag 1 gives us lag 10
res <- data %>% group_by(directory, selection_name) %>%
summarise(
fit_phylo_10 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10
discretize(lag(max_phenotype_pairwise_distance, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_phylo_100 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100
discretize(lag(max_phenotype_pairwise_distance, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_phylo_1000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 1000
discretize(lag(max_phenotype_pairwise_distance, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_phylo_10000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10000
discretize(lag(max_phenotype_pairwise_distance, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_phylo_100000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100000
discretize(lag(max_phenotype_pairwise_distance, 10000)),
discretize(lag(elite_trait_avg, 10000))),
fit_fit_10000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 1000)),
discretize(lag(max_phenotype_pairwise_distance, 1000))),
fit_fit_100000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 10000)),
discretize(lag(max_phenotype_pairwise_distance, 10000))),
fit_pheno_10 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10
discretize(lag(phen_num_taxa, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_pheno_100 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100
discretize(lag(phen_num_taxa, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_pheno_1000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 1000
discretize(lag(phen_num_taxa, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_pheno_10000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10000
discretize(lag(phen_num_taxa, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_pheno_100000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100000
discretize(lag(phen_num_taxa, 10000)),
discretize(lag(elite_trait_avg, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("o_10"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo") ~ "Phylogenetic", TRUE ~ "Phenotypic")
# Plot transfer entropy
ggplot(
res %>% filter(str_detect(name, "fit_ph*")),
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")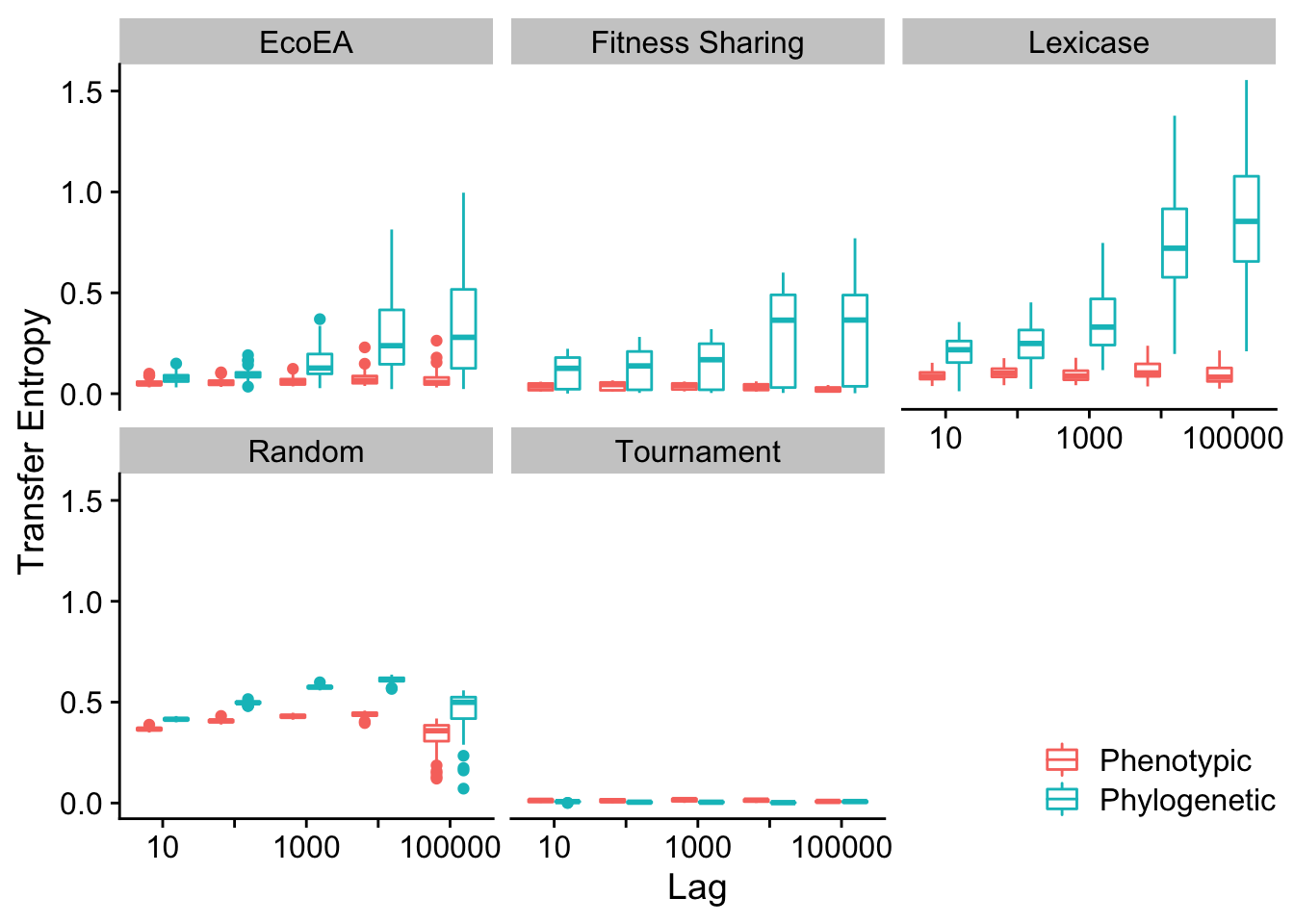
Next we calculate statistics to quantify these differences
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 466 | 1.00e-07 | 0.0000017 | **** | p < 1e-04 | 0.5404755 | large |
| EcoEA | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 234 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7004121 | large |
| EcoEA | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 192 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7293661 | large |
| EcoEA | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 165 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7479795 | large |
| EcoEA | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 242 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6948970 | large |
| Fitness Sharing | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 798 | 1.85e-03 | 0.0462500 |
|
p = 0.04625 | 0.3116007 | moderate |
| Fitness Sharing | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 759 | 7.21e-04 | 0.0180250 |
|
p = 0.018025 | 0.3384866 | moderate |
| Fitness Sharing | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 826 | 3.51e-03 | 0.0877500 | ns | p = 0.08775 | 0.2922980 | small |
| Fitness Sharing | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 661 | 4.97e-05 | 0.0012425 | ** | p = 0.0012425 | 0.4060460 | moderate |
| Fitness Sharing | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 436 | 0.00e+00 | 0.0000005 | **** | p < 1e-04 | 0.5611569 | large |
| Lexicase | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 195 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7272980 | large |
| Lexicase | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 173 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7424644 | large |
| Lexicase | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 34 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8382885 | large |
| Lexicase | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 6 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8575912 | large |
| Lexicase | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8610381 | large |
| Random | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 358 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6149287 | large |
| Tournament | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 1810 | 1.15e-04 | 0.0028750 | ** | p = 0.002875 | 0.3860539 | moderate |
| Tournament | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 2024 | 1.00e-07 | 0.0000024 | **** | p < 1e-04 | 0.5335817 | large |
| Tournament | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 2165 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6307845 | large |
| Tournament | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 2322 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7390175 | large |
| Tournament | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1336 | 5.56e-01 | 1.0000000 | ns | p = 1 | 0.0592869 | small |
2.7.2.2 Mean pairwise distance vs. phenotypic richness
# Calculate transfer entropy for mean pairwise distance
# Time points are 10 generations, so calculating lag 1 gives us lag 10
res <- data %>% group_by(directory, selection_name) %>%
summarise(
fit_phylo_10 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10
discretize(lag(mean_phenotype_pairwise_distance, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_phylo_100 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100
discretize(lag(mean_phenotype_pairwise_distance, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_phylo_1000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 1000
discretize(lag(mean_phenotype_pairwise_distance, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_phylo_10000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10000
discretize(lag(mean_phenotype_pairwise_distance, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_phylo_100000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100000
discretize(lag(mean_phenotype_pairwise_distance, 10000)),
discretize(lag(elite_trait_avg, 10000))),
fit_fit_10000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 1000)),
discretize(lag(mean_phenotype_pairwise_distance, 1000))),
fit_fit_100000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 10000)),
discretize(lag(mean_phenotype_pairwise_distance, 10000))),
fit_pheno_10 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10
discretize(lag(phen_num_taxa, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_pheno_100 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100
discretize(lag(phen_num_taxa, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_pheno_1000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 1000
discretize(lag(phen_num_taxa, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_pheno_10000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10000
discretize(lag(phen_num_taxa, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_pheno_100000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100000
discretize(lag(phen_num_taxa, 10000)),
discretize(lag(elite_trait_avg, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("o_10"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo") ~ "Phylogenetic", TRUE ~ "Phenotypic")
# Plot transfer entropy
ggplot(
res %>% filter(str_detect(name, "fit_ph*")),
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")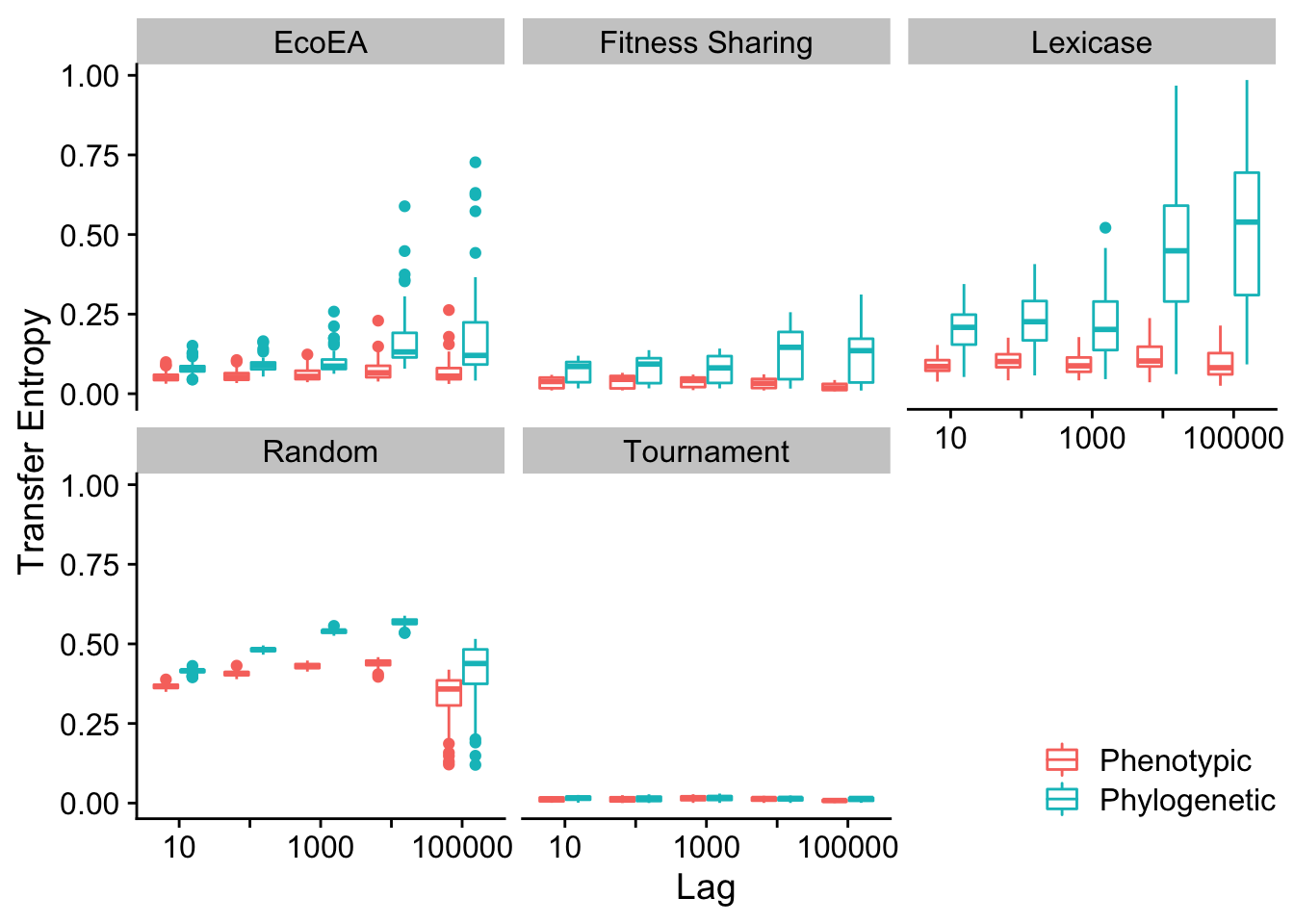
Mean and max pairwise distance appear to behave virtually identically.
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 288 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6631855 | large |
| EcoEA | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 256 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6852457 | large |
| EcoEA | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 330 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6342314 | large |
| EcoEA | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 209 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7176466 | large |
| EcoEA | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 381 | 0.00e+00 | 0.0000001 | **** | p < 1e-04 | 0.5990729 | large |
| Fitness Sharing | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 521 | 5.00e-07 | 0.0000128 | **** | p < 1e-04 | 0.5025595 | large |
| Fitness Sharing | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 550 | 1.40e-06 | 0.0000355 | **** | p < 1e-04 | 0.4825674 | moderate |
| Fitness Sharing | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 612 | 1.11e-05 | 0.0002775 | *** | p = 0.0002775 | 0.4398257 | moderate |
| Fitness Sharing | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 459 | 1.00e-07 | 0.0000013 | **** | p < 1e-04 | 0.5453012 | large |
| Fitness Sharing | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 350 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6204438 | large |
| Lexicase | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 195 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7272980 | large |
| Lexicase | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 220 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7100634 | large |
| Lexicase | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 333 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6321633 | large |
| Lexicase | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 142 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7638352 | large |
| Lexicase | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 115 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7824486 | large |
| Random | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 538 | 9.00e-07 | 0.0000233 | **** | p < 1e-04 | 0.4908400 | moderate |
| Tournament | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 928 | 2.67e-02 | 0.6675000 | ns | p = 0.6675 | 0.2219810 | small |
| Tournament | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 1105 | 3.19e-01 | 1.0000000 | ns | p = 1 | 0.0999604 | small |
| Tournament | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1184 | 6.52e-01 | 1.0000000 | ns | p = 1 | 0.0454992 | small |
| Tournament | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1188 | 6.72e-01 | 1.0000000 | ns | p = 1 | 0.0427417 | small |
| Tournament | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 618 | 1.34e-05 | 0.0003350 | *** | p = 0.000335 | 0.4356894 | moderate |
2.7.2.3 Mean pairwise distance vs. shannon entropy
# Calculate transfer entropy for mean pairwise distance
# Time points are 10 generations, so calculating lag 1 gives us lag 10
res <- data %>% group_by(directory, selection_name) %>%
summarise(
fit_phylo_10 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10
discretize(lag(mean_phenotype_pairwise_distance, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_phylo_100 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100
discretize(lag(mean_phenotype_pairwise_distance, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_phylo_1000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 1000
discretize(lag(mean_phenotype_pairwise_distance, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_phylo_10000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10000
discretize(lag(mean_phenotype_pairwise_distance, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_phylo_100000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100000
discretize(lag(mean_phenotype_pairwise_distance, 10000)),
discretize(lag(elite_trait_avg, 10000))),
fit_fit_10000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 1000)),
discretize(lag(mean_phenotype_pairwise_distance, 1000))),
fit_fit_100000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 10000)),
discretize(lag(mean_phenotype_pairwise_distance, 10000))),
fit_pheno_10 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10
discretize(lag(phen_diversity, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_pheno_100 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100
discretize(lag(phen_diversity, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_pheno_1000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 1000
discretize(lag(phen_diversity, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_pheno_10000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10000
discretize(lag(phen_diversity, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_pheno_100000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100000
discretize(lag(phen_diversity, 10000)),
discretize(lag(elite_trait_avg, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("o_10"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo") ~ "Phylogenetic", TRUE ~ "Phenotypic")
# Plot transfer entropy
ggplot(
res %>% filter(str_detect(name, "fit_ph*")),
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")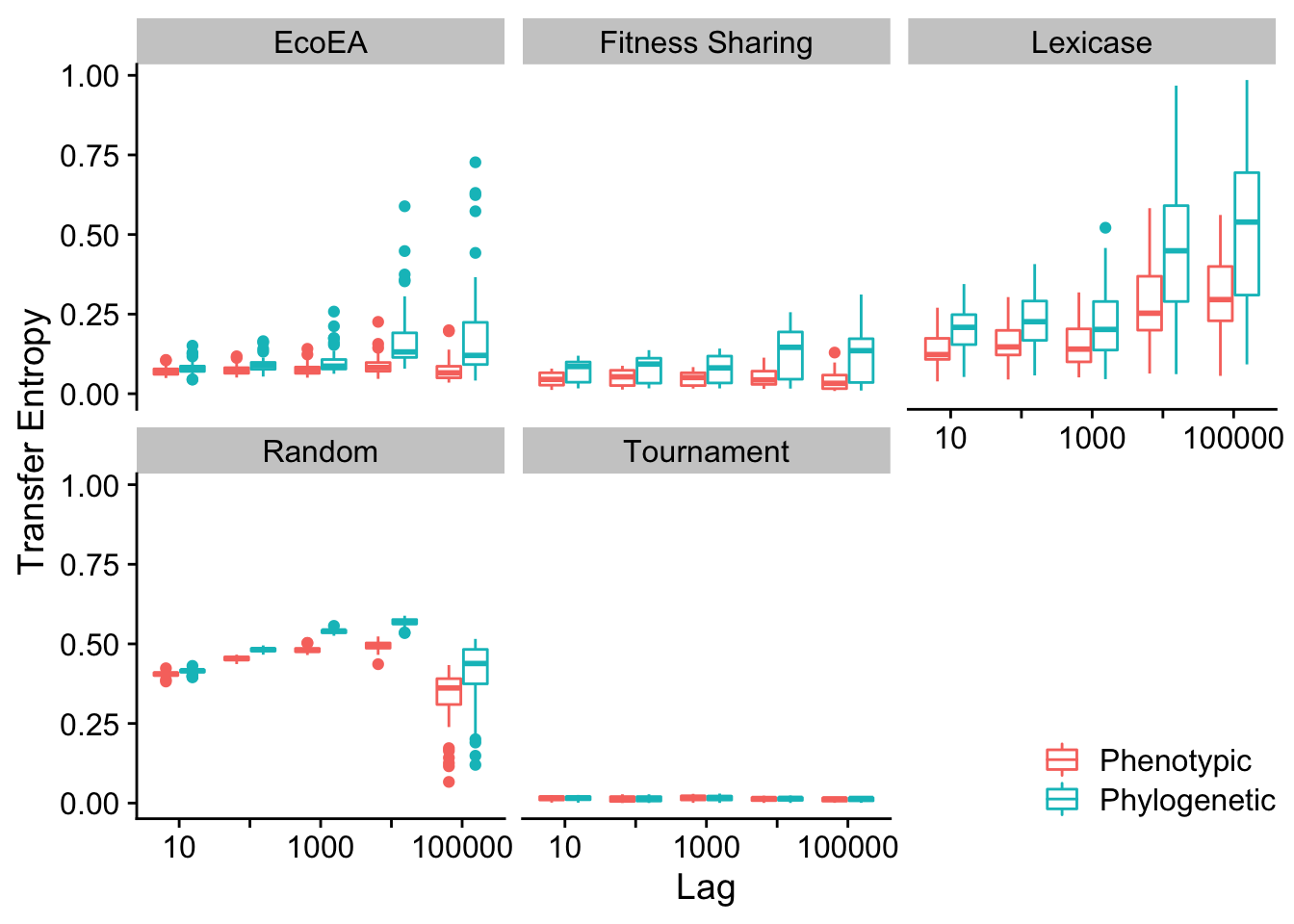
Looks like Shannon Diversity is more predictive of fitness than phenotypic richness (at least for lexicase), although still not as much as mean pairwise distance.
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 789 | 1.50e-03 | 0.0375000 |
|
p = 0.0375 | 0.3178051 | moderate |
| EcoEA | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 666 | 5.76e-05 | 0.0014400 | ** | p = 0.00144 | 0.4025991 | moderate |
| EcoEA | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 678 | 8.15e-05 | 0.0020375 | ** | p = 0.0020375 | 0.3943265 | moderate |
| EcoEA | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 291 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6611173 | large |
| EcoEA | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 445 | 0.00e+00 | 0.0000007 | **** | p < 1e-04 | 0.5549525 | large |
| Fitness Sharing | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 706 | 1.79e-04 | 0.0044750 | ** | p = 0.004475 | 0.3750238 | moderate |
| Fitness Sharing | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 726 | 3.07e-04 | 0.0076750 | ** | p = 0.007675 | 0.3612362 | moderate |
| Fitness Sharing | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 781 | 1.24e-03 | 0.0310000 |
|
p = 0.031 | 0.3233202 | moderate |
| Fitness Sharing | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 617 | 1.30e-05 | 0.0003250 | *** | p = 0.000325 | 0.4363788 | moderate |
| Fitness Sharing | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 538 | 9.00e-07 | 0.0000233 | **** | p < 1e-04 | 0.4908400 | moderate |
| Lexicase | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 561 | 2.10e-06 | 0.0000518 | **** | p < 1e-04 | 0.4749842 | moderate |
| Lexicase | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 612 | 1.11e-05 | 0.0002775 | *** | p = 0.0002775 | 0.4398257 | moderate |
| Lexicase | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 829 | 3.75e-03 | 0.0937500 | ns | p = 0.09375 | 0.2902298 | small |
| Lexicase | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 610 | 1.04e-05 | 0.0002600 | *** | p = 0.00026 | 0.4412045 | moderate |
| Lexicase | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 611 | 1.07e-05 | 0.0002675 | *** | p = 0.0002675 | 0.4405151 | moderate |
| Random | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 404 | 0.00e+00 | 0.0000001 | **** | p < 1e-04 | 0.5832172 | large |
| Random | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 1 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8610381 | large |
| Random | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 580 | 3.90e-06 | 0.0000980 | **** | p < 1e-04 | 0.4618859 | moderate |
| Tournament | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 1241 | 9.53e-01 | 1.0000000 | ns | p = 1 | 0.0062044 | small |
| Tournament | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 1225 | 8.66e-01 | 1.0000000 | ns | p = 1 | 0.0172345 | small |
| Tournament | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1265 | 9.20e-01 | 1.0000000 | ns | p = 1 | 0.0103407 | small |
| Tournament | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1217 | 8.23e-01 | 1.0000000 | ns | p = 1 | 0.0227496 | small |
| Tournament | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1105 | 3.19e-01 | 1.0000000 | ns | p = 1 | 0.0999604 | small |
2.7.2.4 Mean evolutionary distinctiveness vs. phenotypic richness
# Calculate transfer entropy for mean evolutionary distinctiveness
# Time points are 10 generations, so calculating lag 1 gives us lag 10
res <- data %>% group_by(directory, selection_name) %>%
summarise(
fit_phylo_10 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_phylo_100 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_phylo_1000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 1000
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_phylo_10000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10000
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_phylo_100000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100000
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000)),
discretize(lag(elite_trait_avg, 10000))),
fit_fit_10000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 1000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000))),
fit_fit_100000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 10000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000))),
fit_pheno_10 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10
discretize(lag(phen_num_taxa, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_pheno_100 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100
discretize(lag(phen_num_taxa, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_pheno_1000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 1000
discretize(lag(phen_num_taxa, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_pheno_10000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10000
discretize(lag(phen_num_taxa, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_pheno_100000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100000
discretize(lag(phen_num_taxa, 10000)),
discretize(lag(elite_trait_avg, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("o_10"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo") ~ "Phylogenetic", TRUE ~ "Phenotypic")
# Plot transfer entropy
ggplot(
res %>% filter(str_detect(name, "fit_ph*")),
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")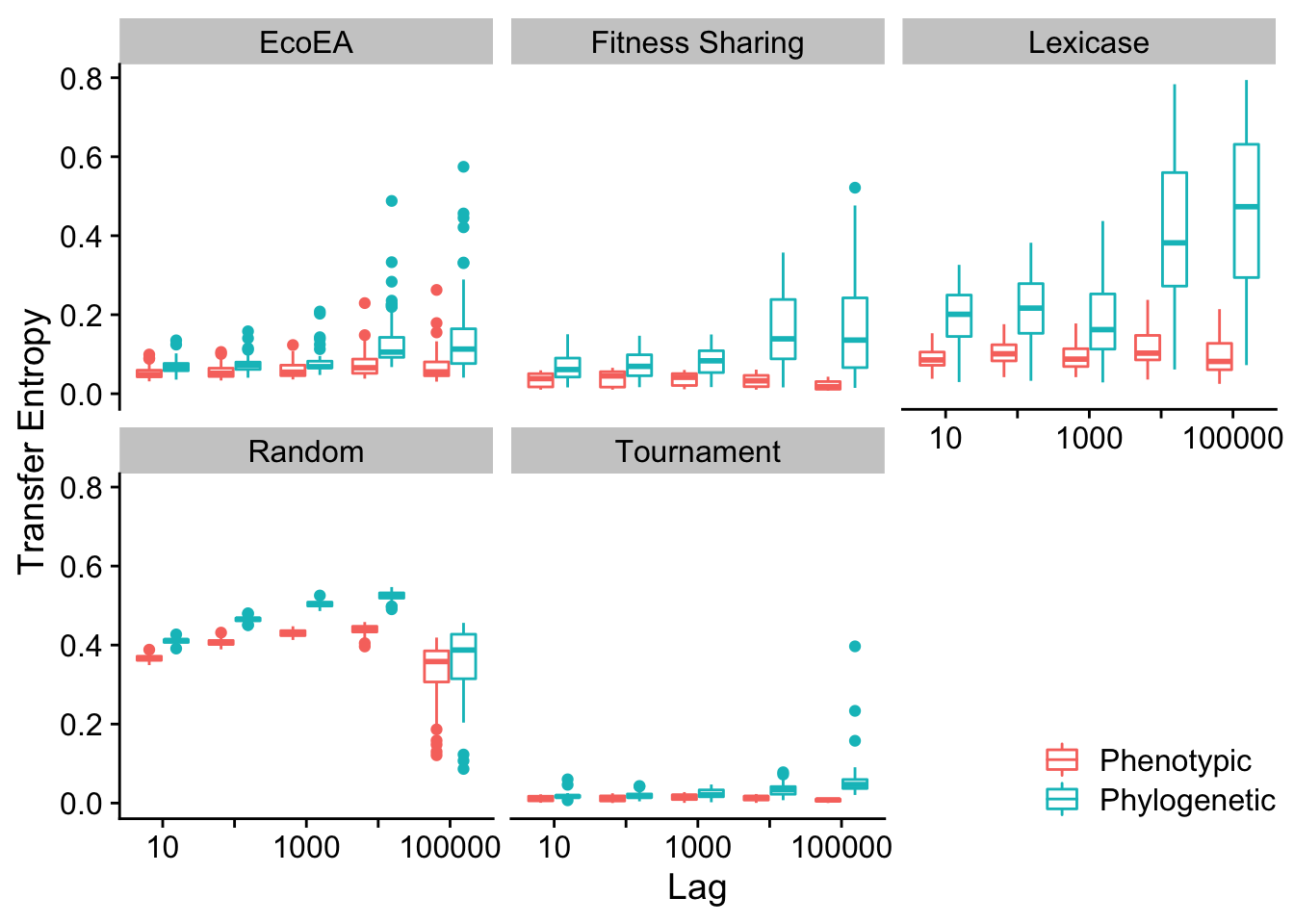
Interestingly, mean evolutionary distinctiveness looks fairly comparable to mean pairwise distance (although note that the y axis doesn’t go quite as far).
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 543 | 1.10e-06 | 0.0000278 | **** | p < 1e-04 | 0.4873931 | moderate |
| EcoEA | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 536 | 9.00e-07 | 0.0000218 | **** | p < 1e-04 | 0.4922187 | moderate |
| EcoEA | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 644 | 2.99e-05 | 0.0007475 | *** | p = 0.0007475 | 0.4177655 | moderate |
| EcoEA | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 380 | 0.00e+00 | 0.0000001 | **** | p < 1e-04 | 0.5997623 | large |
| EcoEA | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 501 | 2.00e-07 | 0.0000062 | **** | p < 1e-04 | 0.5163471 | large |
| Fitness Sharing | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 531 | 7.00e-07 | 0.0000182 | **** | p < 1e-04 | 0.4956656 | moderate |
| Fitness Sharing | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 551 | 1.50e-06 | 0.0000368 | **** | p < 1e-04 | 0.4818780 | moderate |
| Fitness Sharing | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 423 | 0.00e+00 | 0.0000003 | **** | p < 1e-04 | 0.5701189 | large |
| Fitness Sharing | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 321 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6404359 | large |
| Fitness Sharing | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 107 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7879636 | large |
| Lexicase | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 237 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6983440 | large |
| Lexicase | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 275 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6721474 | large |
| Lexicase | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 516 | 4.00e-07 | 0.0000107 | **** | p < 1e-04 | 0.5060064 | large |
| Lexicase | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 220 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7100634 | large |
| Lexicase | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 178 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7390175 | large |
| Random | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 893 | 1.40e-02 | 0.3500000 | ns | p = 0.35 | 0.2461094 | small |
| Tournament | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 654 | 4.04e-05 | 0.0010100 | ** | p = 0.00101 | 0.4108717 | moderate |
| Tournament | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 718 | 2.48e-04 | 0.0062000 | ** | p = 0.0062 | 0.3667512 | moderate |
| Tournament | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 735 | 3.90e-04 | 0.0097500 | ** | p = 0.00975 | 0.3550317 | moderate |
| Tournament | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 180 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7376387 | large |
| Tournament | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
2.7.2.5 Mean evolutionary distinctiveness vs. shannon entropy
# Calculate transfer entropy for mean evolutionary distinctiveness
# Time points are 10 generations, so calculating lag 1 gives us lag 10
res <- data %>% group_by(directory, selection_name) %>%
summarise(
fit_phylo_10 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_phylo_100 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_phylo_1000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 1000
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_phylo_10000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 10000
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_phylo_100000 = condinformation(discretize(elite_trait_avg), # TE phylo -> fit, lag 100000
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000)),
discretize(lag(elite_trait_avg, 10000))),
fit_fit_10000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 1000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000))),
fit_fit_100000 = condinformation(discretize(elite_trait_avg), # Mutual info btwn. fit and lagged fit
discretize(lag(elite_trait_avg, 10000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000))),
fit_pheno_10 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10
discretize(lag(phen_diversity, 1)),
discretize(lag(elite_trait_avg, 1))),
fit_pheno_100 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100
discretize(lag(phen_diversity, 10)),
discretize(lag(elite_trait_avg, 10))),
fit_pheno_1000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 1000
discretize(lag(phen_diversity, 100)),
discretize(lag(elite_trait_avg, 100))),
fit_pheno_10000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 10000
discretize(lag(phen_diversity, 1000)),
discretize(lag(elite_trait_avg, 1000))),
fit_pheno_100000 = condinformation(discretize(elite_trait_avg), # TE pheno -> fit, lag 100000
discretize(lag(phen_diversity, 10000)),
discretize(lag(elite_trait_avg, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("o_10"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo") ~ "Phylogenetic", TRUE ~ "Phenotypic")
# Plot transfer entropy
ggplot(
res %>% filter(str_detect(name, "fit_ph*")),
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")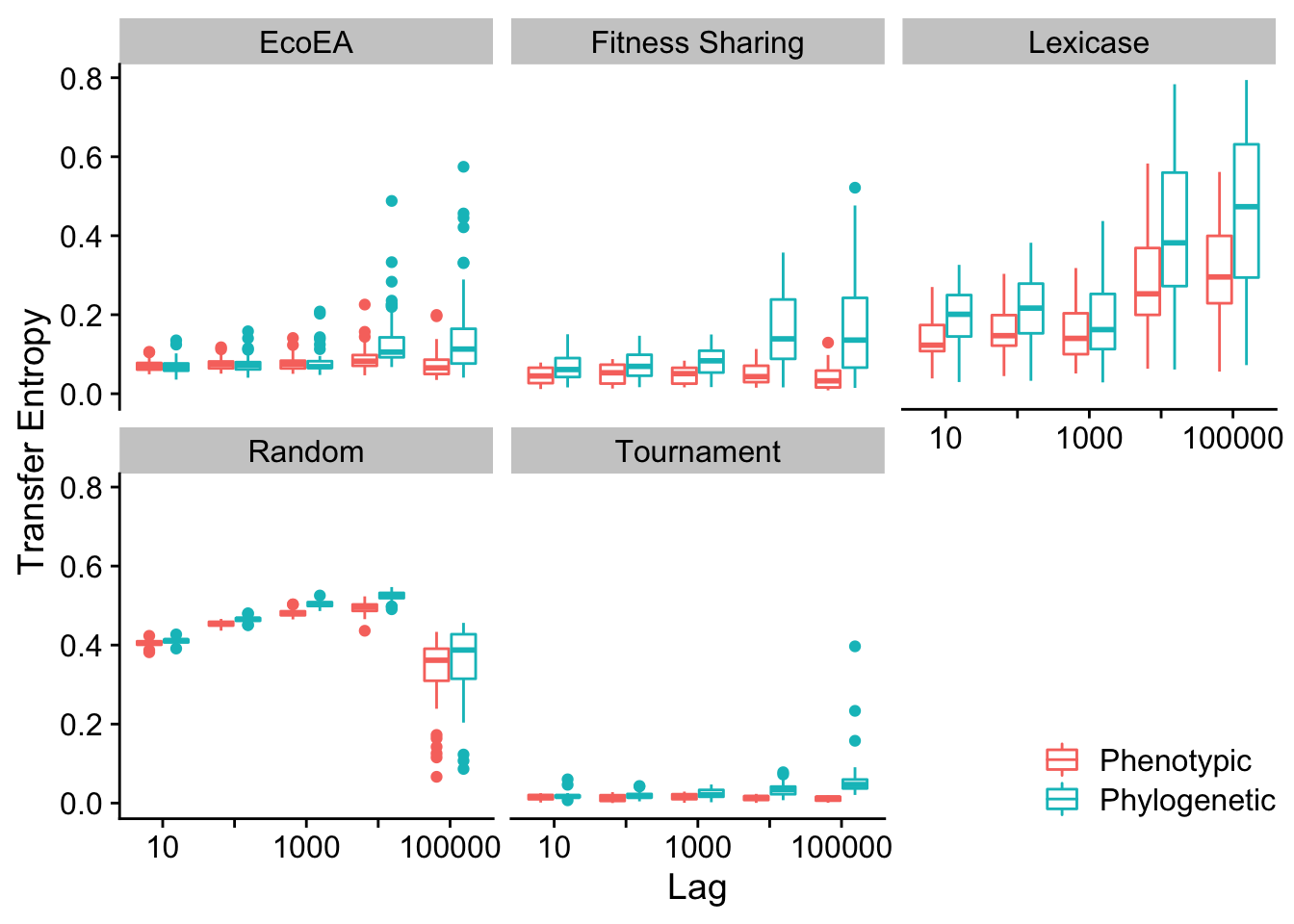
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 1381 | 3.68e-01 | 1.0000000 | ns | p = 1 | 0.0903090 | small |
| EcoEA | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 1356 | 4.67e-01 | 1.0000000 | ns | p = 1 | 0.0730745 | small |
| EcoEA | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1345 | 5.15e-01 | 1.0000000 | ns | p = 1 | 0.0654913 | small |
| EcoEA | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 527 | 6.00e-07 | 0.0000158 | **** | p < 1e-04 | 0.4984232 | moderate |
| EcoEA | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 585 | 4.60e-06 | 0.0001158 | *** | p = 0.00011575 | 0.4584390 | moderate |
| Fitness Sharing | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 816 | 2.80e-03 | 0.0700000 | ns | p = 0.07 | 0.2991918 | small |
| Fitness Sharing | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 839 | 4.66e-03 | 0.1165000 | ns | p = 0.1165 | 0.2833360 | small |
| Fitness Sharing | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 620 | 1.43e-05 | 0.0003575 | *** | p = 0.0003575 | 0.4343107 | moderate |
| Fitness Sharing | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 452 | 0.00e+00 | 0.0000010 | **** | p < 1e-04 | 0.5501268 | large |
| Fitness Sharing | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 328 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6356102 | large |
| Lexicase | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 644 | 2.99e-05 | 0.0007475 | *** | p = 0.0007475 | 0.4177655 | moderate |
| Lexicase | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 700 | 1.52e-04 | 0.0038000 | ** | p = 0.0038 | 0.3791601 | moderate |
| Lexicase | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 1043 | 1.55e-01 | 1.0000000 | ns | p = 1 | 0.1427021 | small |
| Lexicase | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 766 | 8.59e-04 | 0.0214750 |
|
p = 0.021475 | 0.3336609 | moderate |
| Lexicase | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 693 | 1.25e-04 | 0.0031250 | ** | p = 0.003125 | 0.3839858 | moderate |
| Random | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 718 | 2.48e-04 | 0.0062000 | ** | p = 0.0062 | 0.3667512 | moderate |
| Random | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 271 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6749050 | large |
| Random | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 72 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8120920 | large |
| Random | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 130 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7721078 | large |
| Random | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 957 | 4.38e-02 | 1.0000000 | ns | p = 1 | 0.2019889 | small |
| Tournament | 10 | value | Phenotypic | Phylogenetic | 50 | 50 | 1034 | 1.37e-01 | 1.0000000 | ns | p = 1 | 0.1489065 | small |
| Tournament | 100 | value | Phenotypic | Phylogenetic | 50 | 50 | 867 | 8.37e-03 | 0.2092500 | ns | p = 0.20925 | 0.2640333 | small |
| Tournament | 1000 | value | Phenotypic | Phylogenetic | 50 | 50 | 791 | 1.57e-03 | 0.0392500 |
|
p = 0.03925 | 0.3164263 | moderate |
| Tournament | 10000 | value | Phenotypic | Phylogenetic | 50 | 50 | 195 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7272980 | large |
| Tournament | 100000 | value | Phenotypic | Phylogenetic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
2.7.3 Transfer entropy between types of diversity
While we’re calculating transfer entropy, we might as well also calculate it between phenotypic diversity and phylogenetic diversity, as these could potentially also be in a feedback loop.
2.7.3.1 Max pairwise distance and phenotypic richness
res <- data %>% group_by(directory, selection_name) %>%
summarise(
phen_phylo_10 = condinformation(discretize(phen_num_taxa),
discretize(lag(max_phenotype_pairwise_distance, 1)),
discretize(lag(phen_num_taxa, 1))),
phen_phylo_100 = condinformation(discretize(phen_num_taxa),
discretize(lag(max_phenotype_pairwise_distance, 10)),
discretize(lag(phen_num_taxa, 10))),
pheno_phylo_1000 = condinformation(discretize(phen_num_taxa),
discretize(lag(max_phenotype_pairwise_distance, 100)),
discretize(lag(phen_num_taxa, 100))),
pheno_phylo_10000 = condinformation(discretize(phen_num_taxa),
discretize(lag(max_phenotype_pairwise_distance, 1000)),
discretize(lag(phen_num_taxa, 1000))),
pheno_phylo_100000 = condinformation(discretize(phen_num_taxa),
discretize(lag(max_phenotype_pairwise_distance, 10000)),
discretize(lag(phen_num_taxa, 10000))),
phylo_pheno_10 = condinformation(discretize(max_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 1)),
discretize(lag(max_phenotype_pairwise_distance, 1))),
phylo_pheno_100 = condinformation(discretize(max_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 10)),
discretize(lag(max_phenotype_pairwise_distance, 10))),
phylo_pheno_1000 = condinformation(discretize(max_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 100)),
discretize(lag(max_phenotype_pairwise_distance, 100))),
phylo_pheno_10000 = condinformation(discretize(max_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 1000)),
discretize(lag(max_phenotype_pairwise_distance, 1000))),
phylo_pheno_100000 = condinformation(discretize(max_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 10000)),
discretize(lag(max_phenotype_pairwise_distance, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("phylo"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo_pheno") ~ "\nPhenotypic\n\t->\nPhylogenetic\n", TRUE ~ "\nPhylogenetic\n\t->\nPhenotypic\n")
ggplot(
res,
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")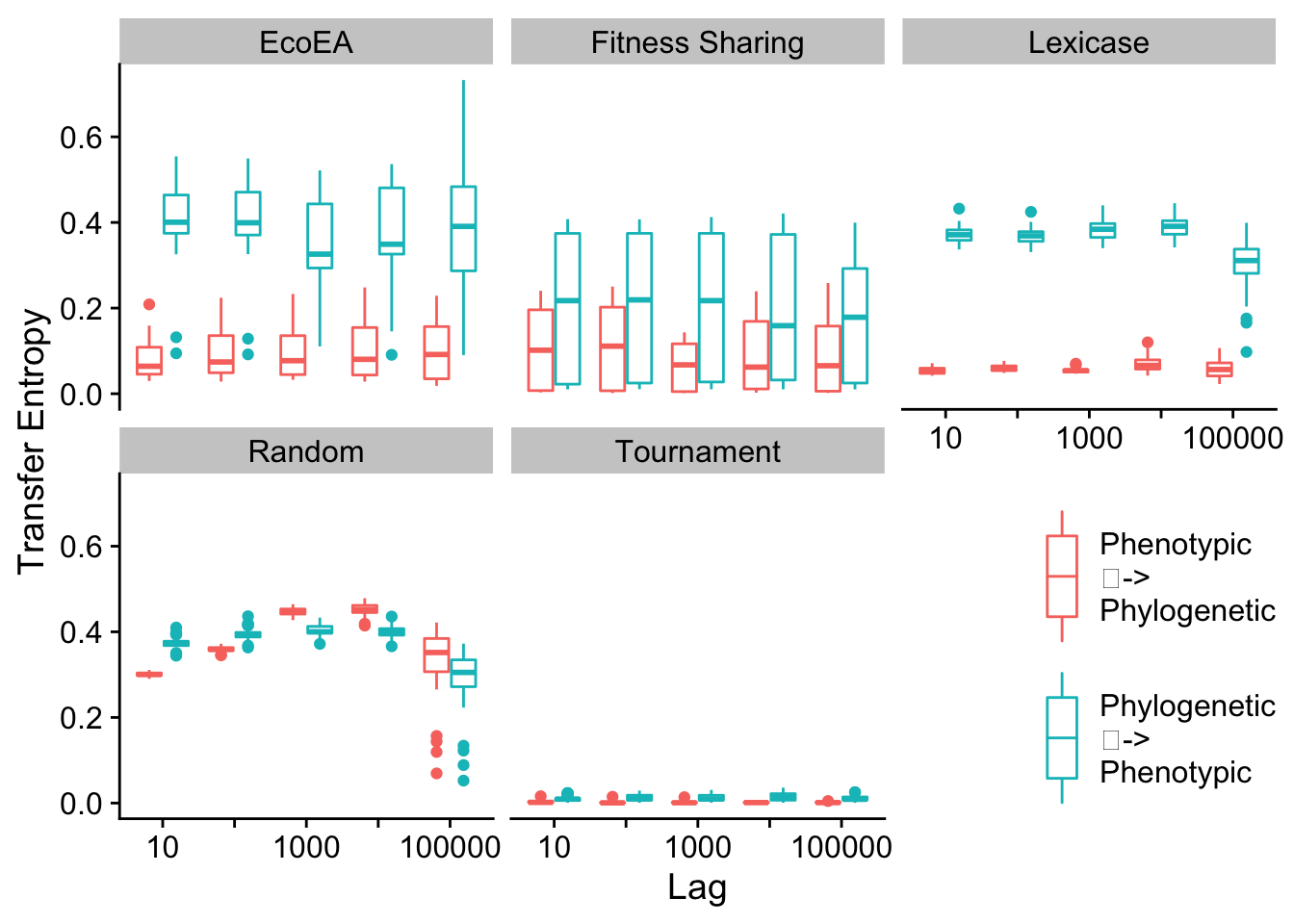
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 23 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8458717 | large |
| EcoEA | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 35 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8375991 | large |
| EcoEA | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 27 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8431142 | large |
| EcoEA | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 38 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8355310 | large |
| EcoEA | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 69 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8141601 | large |
| Fitness Sharing | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 768 | 9.02e-04 | 0.0225500 |
|
p = 0.02255 | 0.3322821 | moderate |
| Fitness Sharing | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 747 | 5.32e-04 | 0.0133000 |
|
p = 0.0133 | 0.3467591 | moderate |
| Fitness Sharing | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 666 | 5.76e-05 | 0.0014400 | ** | p = 0.00144 | 0.4025991 | moderate |
| Fitness Sharing | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 732 | 3.60e-04 | 0.0090000 | ** | p = 0.009 | 0.3570999 | moderate |
| Fitness Sharing | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 745 | 5.05e-04 | 0.0126250 |
|
p = 0.012625 | 0.3481379 | moderate |
| Lexicase | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8610381 | large |
| Random | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 15 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8513868 | large |
| Random | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2497 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8596593 | large |
| Random | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2479 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8472505 | large |
| Random | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1849 | 3.69e-05 | 0.0009225 | *** | p = 0.0009225 | 0.4129398 | moderate |
| Tournament | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 168 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7459113 | large |
| Tournament | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 53 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8251902 | large |
| Tournament | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 74 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8107132 | large |
| Tournament | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 60 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8203646 | large |
| Tournament | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 115 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7824486 | large |
2.7.3.2 Mean pairwise distance and phenotypic richness
res <- data %>% group_by(directory, selection_name) %>%
summarise(
phen_phylo_10 = condinformation(discretize(phen_num_taxa),
discretize(lag(mean_phenotype_pairwise_distance, 1)),
discretize(lag(phen_num_taxa, 1))),
phen_phylo_100 = condinformation(discretize(phen_num_taxa),
discretize(lag(mean_phenotype_pairwise_distance, 10)),
discretize(lag(phen_num_taxa, 10))),
pheno_phylo_1000 = condinformation(discretize(phen_num_taxa),
discretize(lag(mean_phenotype_pairwise_distance, 100)),
discretize(lag(phen_num_taxa, 100))),
pheno_phylo_10000 = condinformation(discretize(phen_num_taxa),
discretize(lag(mean_phenotype_pairwise_distance, 1000)),
discretize(lag(phen_num_taxa, 1000))),
pheno_phylo_100000 = condinformation(discretize(phen_num_taxa),
discretize(lag(mean_phenotype_pairwise_distance, 10000)),
discretize(lag(phen_num_taxa, 10000))),
phylo_pheno_10 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 1)),
discretize(lag(mean_phenotype_pairwise_distance, 1))),
phylo_pheno_100 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 10)),
discretize(lag(mean_phenotype_pairwise_distance, 10))),
phylo_pheno_1000 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 100)),
discretize(lag(mean_phenotype_pairwise_distance, 100))),
phylo_pheno_10000 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 1000)),
discretize(lag(mean_phenotype_pairwise_distance, 1000))),
phylo_pheno_100000 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_num_taxa, 10000)),
discretize(lag(mean_phenotype_pairwise_distance, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("phylo"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo_pheno") ~ "\nPhenotypic\n\t->\nPhylogenetic\n", TRUE ~ "\nPhylogenetic\n\t->\nPhenotypic\n")
ggplot(
res,
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")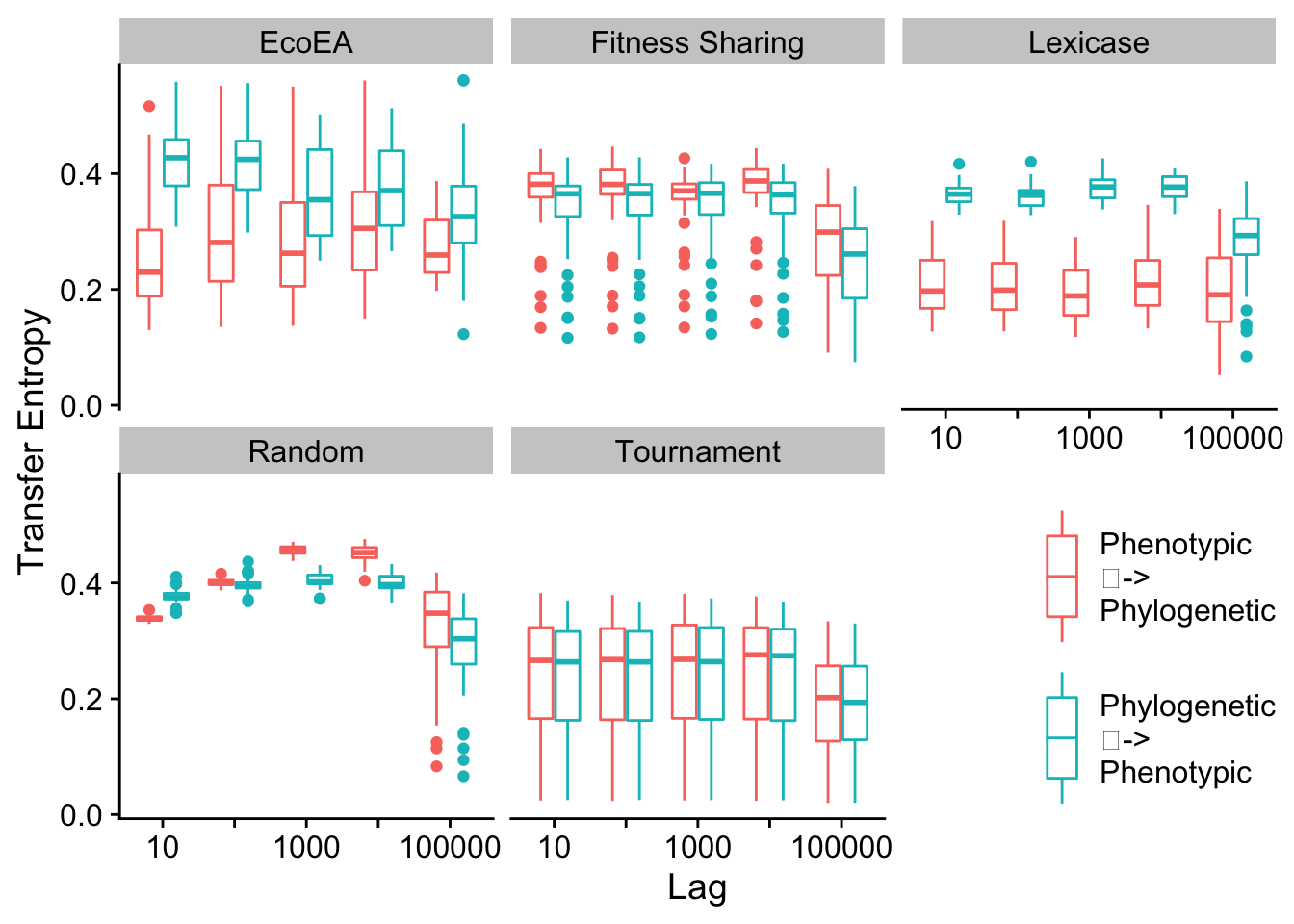
Here, on the other hand, there’s a pretty notable difference between mean and max pairwise distance. Looks like max pairwise distance is in general more predictive of phenotypic richness than mean pairwise distance is (for fitness sharing, mean richness even has a high transfer entropy to MPD than the other way around).
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 342 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6259588 | large |
| EcoEA | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 488 | 2.00e-07 | 0.0000038 | **** | p < 1e-04 | 0.5253091 | large |
| EcoEA | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 661 | 4.97e-05 | 0.0012425 | ** | p = 0.0012425 | 0.4060460 | moderate |
| EcoEA | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 720 | 2.62e-04 | 0.0065500 | ** | p = 0.00655 | 0.3653725 | moderate |
| EcoEA | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 735 | 3.90e-04 | 0.0097500 | ** | p = 0.00975 | 0.3550317 | moderate |
| Fitness Sharing | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1602 | 1.54e-02 | 0.3850000 | ns | p = 0.385 | 0.2426625 | small |
| Fitness Sharing | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1653 | 5.52e-03 | 0.1380000 | ns | p = 0.138 | 0.2778209 | small |
| Fitness Sharing | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1369 | 4.14e-01 | 1.0000000 | ns | p = 1 | 0.0820365 | small |
| Fitness Sharing | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1744 | 6.69e-04 | 0.0167250 |
|
p = 0.016725 | 0.3405547 | moderate |
| Fitness Sharing | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1571 | 2.71e-02 | 0.6775000 | ns | p = 0.6775 | 0.2212916 | small |
| Lexicase | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 4 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8589700 | large |
| Lexicase | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 409 | 0.00e+00 | 0.0000002 | **** | p < 1e-04 | 0.5797703 | large |
| Random | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8603487 | large |
| Random | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1771 | 3.33e-04 | 0.0083250 | ** | p = 0.008325 | 0.3591680 | moderate |
| Random | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2500 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2473 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8431142 | large |
| Random | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1749 | 5.89e-04 | 0.0147250 |
|
p = 0.014725 | 0.3440016 | moderate |
| Tournament | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1298 | 7.43e-01 | 1.0000000 | ns | p = 1 | 0.0330903 | small |
| Tournament | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1306 | 7.02e-01 | 1.0000000 | ns | p = 1 | 0.0386054 | small |
| Tournament | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1301 | 7.28e-01 | 1.0000000 | ns | p = 1 | 0.0351585 | small |
| Tournament | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1276 | 8.60e-01 | 1.0000000 | ns | p = 1 | 0.0179239 | small |
| Tournament | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1280 | 8.39e-01 | 1.0000000 | ns | p = 1 | 0.0206815 | small |
2.7.3.3 Mean pairwise distance and shannon diversity
res <- data %>% group_by(directory, selection_name) %>%
summarise(
phen_phylo_10 = condinformation(discretize(phen_diversity),
discretize(lag(mean_phenotype_pairwise_distance, 1)),
discretize(lag(phen_diversity, 1))),
phen_phylo_100 = condinformation(discretize(phen_diversity),
discretize(lag(mean_phenotype_pairwise_distance, 10)),
discretize(lag(phen_diversity, 10))),
pheno_phylo_1000 = condinformation(discretize(phen_diversity),
discretize(lag(mean_phenotype_pairwise_distance, 100)),
discretize(lag(phen_diversity, 100))),
pheno_phylo_10000 = condinformation(discretize(phen_diversity),
discretize(lag(mean_phenotype_pairwise_distance, 1000)),
discretize(lag(phen_diversity, 1000))),
pheno_phylo_100000 = condinformation(discretize(phen_diversity),
discretize(lag(mean_phenotype_pairwise_distance, 10000)),
discretize(lag(phen_diversity, 10000))),
phylo_pheno_10 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_diversity, 1)),
discretize(lag(mean_phenotype_pairwise_distance, 1))),
phylo_pheno_100 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_diversity, 10)),
discretize(lag(mean_phenotype_pairwise_distance, 10))),
phylo_pheno_1000 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_diversity, 100)),
discretize(lag(mean_phenotype_pairwise_distance, 100))),
phylo_pheno_10000 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_diversity, 1000)),
discretize(lag(mean_phenotype_pairwise_distance, 1000))),
phylo_pheno_100000 = condinformation(discretize(mean_phenotype_pairwise_distance),
discretize(lag(phen_diversity, 10000)),
discretize(lag(mean_phenotype_pairwise_distance, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("phylo"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo_pheno") ~ "\nPhenotypic\n\t->\nPhylogenetic\n", TRUE ~ "\nPhylogenetic\n\t->\nPhenotypic\n")
ggplot(
res,
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")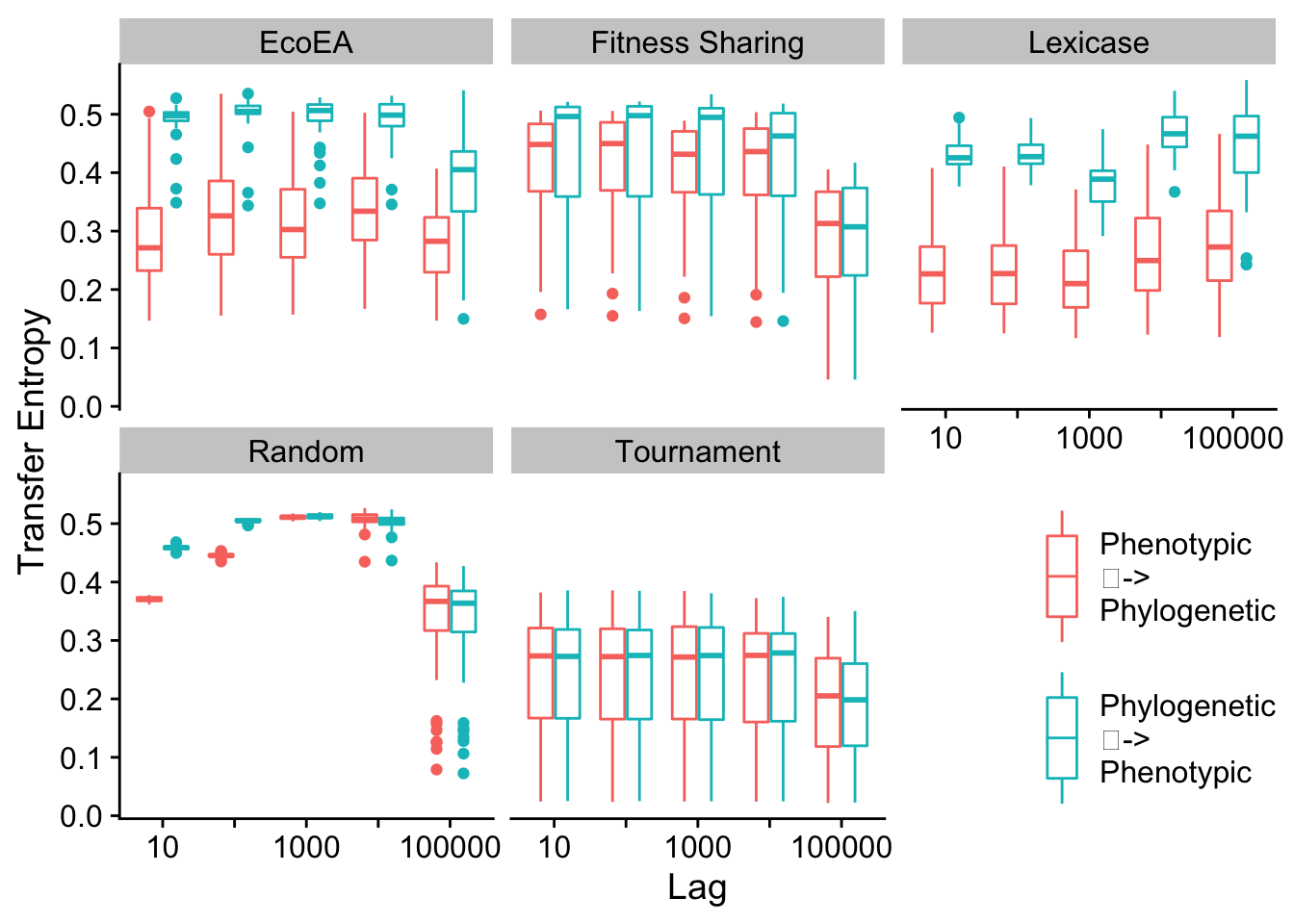
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 101 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7920999 | large |
| EcoEA | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 176 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7403963 | large |
| EcoEA | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 91 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7989937 | large |
| EcoEA | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 119 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7796910 | large |
| EcoEA | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 473 | 1.00e-07 | 0.0000022 | **** | p < 1e-04 | 0.5356498 | large |
| Fitness Sharing | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 884 | 1.17e-02 | 0.2925000 | ns | p = 0.2925 | 0.2523138 | small |
| Fitness Sharing | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 905 | 1.76e-02 | 0.4400000 | ns | p = 0.44 | 0.2378368 | small |
| Fitness Sharing | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 836 | 4.36e-03 | 0.1090000 | ns | p = 0.109 | 0.2854041 | small |
| Fitness Sharing | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 994 | 7.82e-02 | 1.0000000 | ns | p = 1 | 0.1764818 | small |
| Fitness Sharing | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1247 | 9.86e-01 | 1.0000000 | ns | p = 1 | 0.0020681 | small |
| Lexicase | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 6 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8575912 | large |
| Lexicase | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 9 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8555230 | large |
| Lexicase | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 44 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8313947 | large |
| Lexicase | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 24 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8451823 | large |
| Lexicase | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 195 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.7272980 | large |
| Random | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 949 | 3.83e-02 | 0.9575000 | ns | p = 0.9575 | 0.2075040 | small |
| Random | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1558 | 3.40e-02 | 0.8500000 | ns | p = 0.85 | 0.2123297 | small |
| Random | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1304 | 7.12e-01 | 1.0000000 | ns | p = 1 | 0.0372266 | small |
| Tournament | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1232 | 9.04e-01 | 1.0000000 | ns | p = 1 | 0.0124089 | small |
| Tournament | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1261 | 9.42e-01 | 1.0000000 | ns | p = 1 | 0.0075832 | small |
| Tournament | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1252 | 9.92e-01 | 1.0000000 | ns | p = 1 | 0.0013788 | small |
| Tournament | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1217 | 8.23e-01 | 1.0000000 | ns | p = 1 | 0.0227496 | small |
| Tournament | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1240 | 9.48e-01 | 1.0000000 | ns | p = 1 | 0.0068938 | small |
2.7.3.4 Mean evolutionary distinctiveness and phenotypic richness
res <- data %>% group_by(directory, selection_name) %>%
summarise(
phen_phylo_10 = condinformation(
discretize(phen_num_taxa),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1)),
discretize(lag(phen_num_taxa, 1))),
phen_phylo_100 = condinformation(
discretize(phen_num_taxa),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10)),
discretize(lag(phen_num_taxa, 10))),
pheno_phylo_1000 = condinformation(
discretize(phen_num_taxa),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 100)),
discretize(lag(phen_num_taxa, 100))),
pheno_phylo_10000 = condinformation(
discretize(phen_num_taxa),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000)),
discretize(lag(phen_num_taxa, 1000))),
pheno_phylo_100000 = condinformation(
discretize(phen_num_taxa),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000)),
discretize(lag(phen_num_taxa, 10000))),
phylo_pheno_10 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_num_taxa, 1)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1))),
phylo_pheno_100 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_num_taxa, 10)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10))),
phylo_pheno_1000 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_num_taxa, 100)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 100))),
phylo_pheno_10000 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_num_taxa, 1000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000))),
phylo_pheno_100000 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_num_taxa, 10000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("phylo"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo_pheno") ~ "\nPhenotypic\n\t->\nPhylogenetic\n", TRUE ~ "\nPhylogenetic\n\t->\nPhenotypic\n")
ggplot(
res,
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")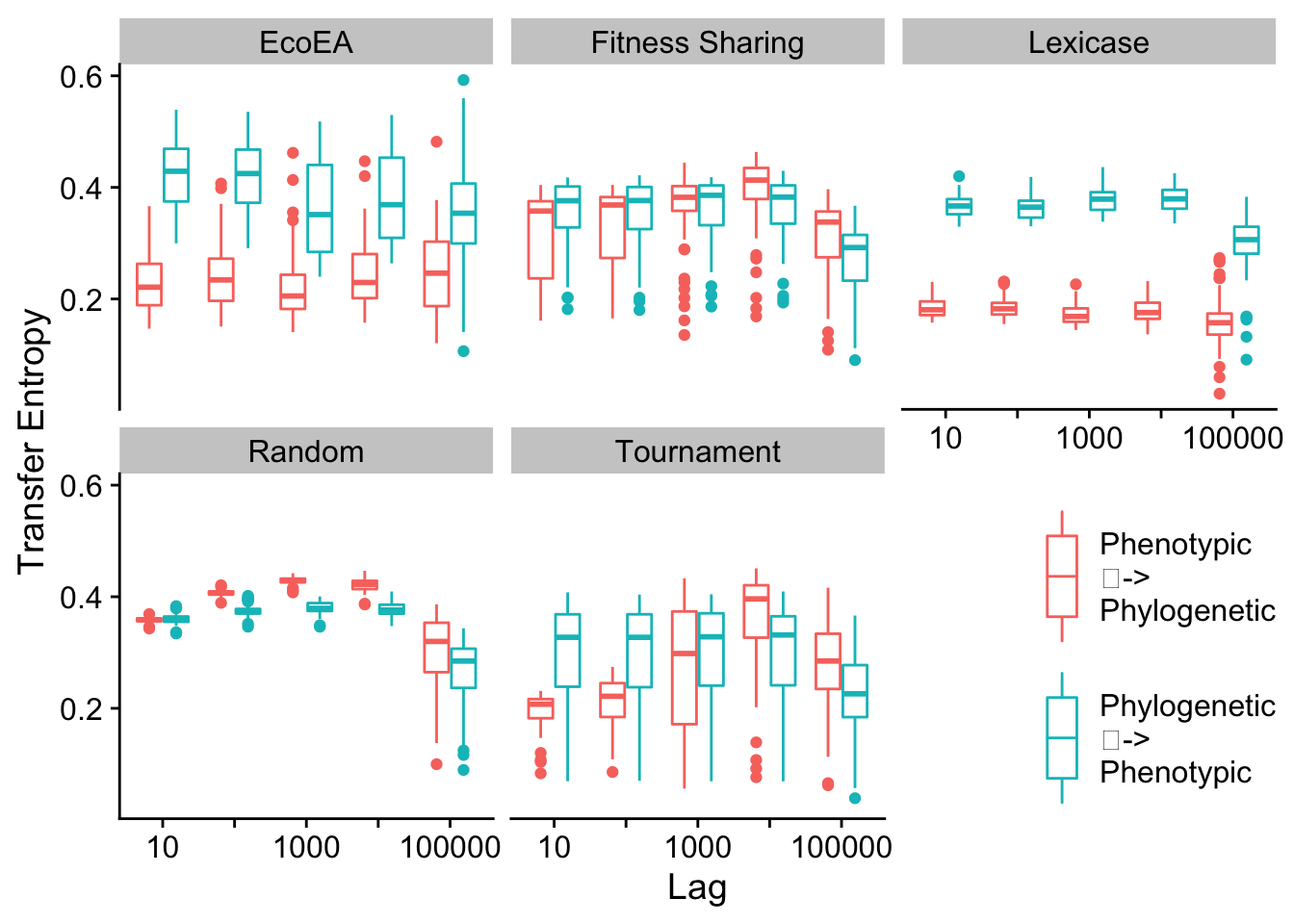
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 25 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8444929 | large |
| EcoEA | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 96 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.7955468 | large |
| EcoEA | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 175 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.7410856 | large |
| EcoEA | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 216 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.7128210 | large |
| EcoEA | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 471 | 1.00e-07 | 0.000002 | **** | p < 1e-04 | 0.5370286 | large |
| Fitness Sharing | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 817 | 2.87e-03 | 0.071750 | ns | p = 0.07175 | 0.2985024 | small |
| Fitness Sharing | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 946 | 3.64e-02 | 0.910000 | ns | p = 0.91 | 0.2095721 | small |
| Fitness Sharing | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1303 | 7.17e-01 | 1.000000 | ns | p = 1 | 0.0365372 | small |
| Fitness Sharing | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1795 | 1.74e-04 | 0.004350 | ** | p = 0.00435 | 0.3757132 | moderate |
| Fitness Sharing | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1693 | 2.28e-03 | 0.057000 | ns | p = 0.057 | 0.3053962 | moderate |
| Lexicase | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 154 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.7555627 | large |
| Random | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1159 | 5.33e-01 | 1.000000 | ns | p = 1 | 0.0627338 | small |
| Random | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2482 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8493186 | large |
| Random | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2500 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2474 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.8438036 | large |
| Random | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1707 | 1.65e-03 | 0.041250 |
|
p = 0.04125 | 0.3150476 | moderate |
| Tournament | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 356 | 0.00e+00 | 0.000000 | **** | p < 1e-04 | 0.6163075 | large |
| Tournament | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 454 | 0.00e+00 | 0.000001 | **** | p < 1e-04 | 0.5487481 | large |
| Tournament | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1154 | 5.10e-01 | 1.000000 | ns | p = 1 | 0.0661807 | small |
| Tournament | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1843 | 4.42e-05 | 0.001105 | ** | p = 0.001105 | 0.4088035 | moderate |
| Tournament | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1736 | 8.17e-04 | 0.020425 |
|
p = 0.020425 | 0.3350396 | moderate |
2.7.3.5 Mean evolutionary distinctiveness and shannon diversity
res <- data %>% group_by(directory, selection_name) %>%
summarise(
phen_phylo_10 = condinformation(
discretize(phen_diversity),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1)),
discretize(lag(phen_diversity, 1))),
phen_phylo_100 = condinformation(
discretize(phen_diversity),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10)),
discretize(lag(phen_diversity, 10))),
pheno_phylo_1000 = condinformation(
discretize(phen_diversity),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 100)),
discretize(lag(phen_diversity, 100))),
pheno_phylo_10000 = condinformation(
discretize(phen_diversity),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000)),
discretize(lag(phen_diversity, 1000))),
pheno_phylo_100000 = condinformation(
discretize(phen_diversity),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000)),
discretize(lag(phen_diversity, 10000))),
phylo_pheno_10 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_diversity, 1)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1))),
phylo_pheno_100 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_diversity, 10)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10))),
phylo_pheno_1000 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_diversity, 100)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 100))),
phylo_pheno_10000 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_diversity, 1000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 1000))),
phylo_pheno_100000 = condinformation(
discretize(mean_phenotype_evolutionary_distinctiveness),
discretize(lag(phen_diversity, 10000)),
discretize(lag(mean_phenotype_evolutionary_distinctiveness, 10000)))
)
# Turn Transfer Entropy columns into rows
res <- res %>% pivot_longer(cols=contains("phylo"))
# Pull lag into a column
res$offset <- str_extract(res$name, "[:digit:]*$")
# Make column indicating direction of transfer entropy
res$Type <- case_when(str_detect(res$name, "phylo_pheno") ~ "\nPhenotypic\n\t->\nPhylogenetic\n", TRUE ~ "\nPhylogenetic\n\t->\nPhenotypic\n")
ggplot(
res,
aes(
x=as.factor(offset),
y=value,
color=Type
)
) +
geom_boxplot() +
facet_wrap(~selection_name) +
scale_x_discrete("Lag",labels=c("10","","1000","","100000")) +
scale_y_continuous("Transfer Entropy") +
theme(legend.position = c(1, 0),
legend.justification = c(1, 0)) +
scale_color_discrete("")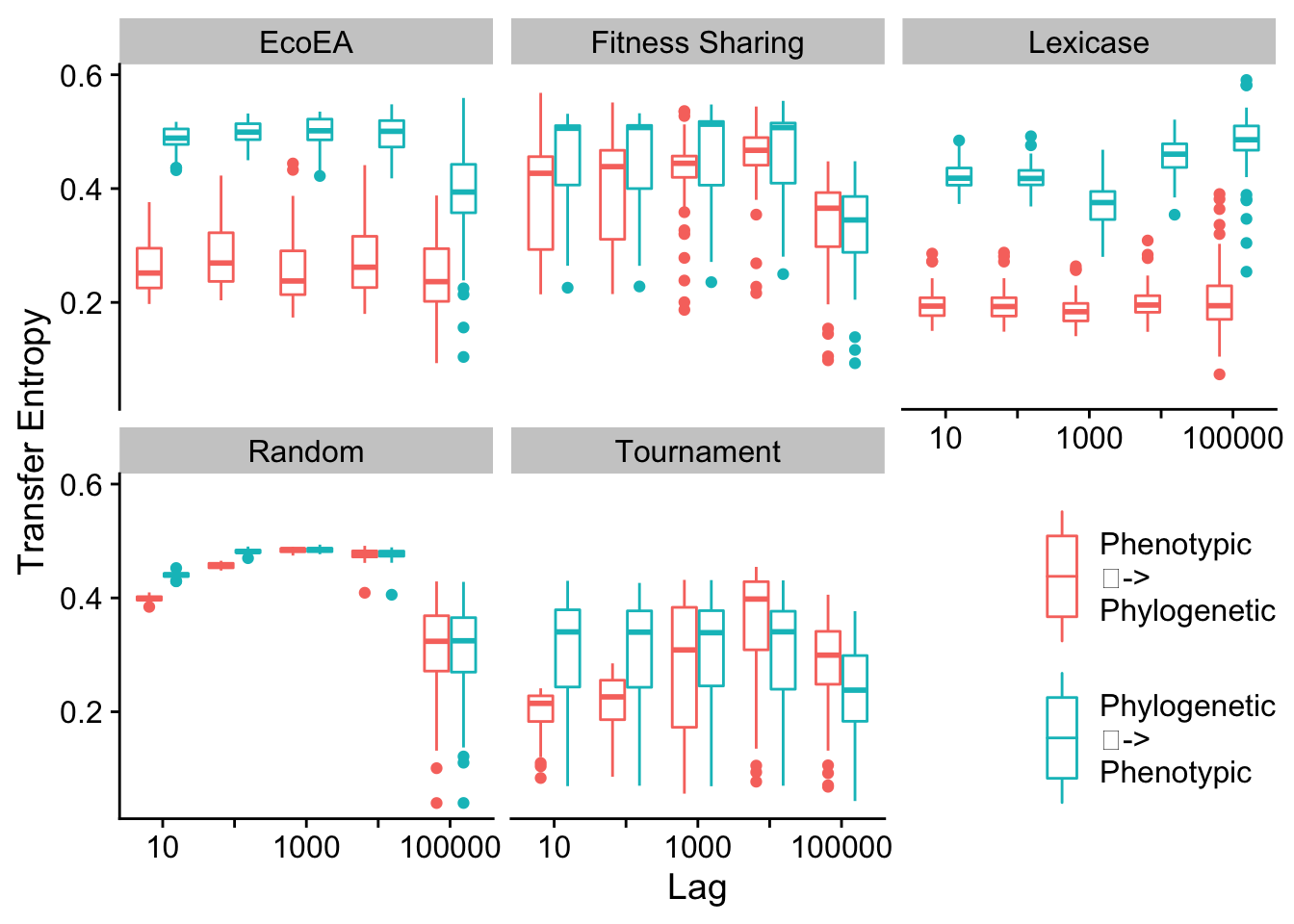
# Determine which conditions are significantly different from each other
transfer_entropy_stats(res)| selection_name | offset | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif | label | effsize | magnitude |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EcoEA | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| EcoEA | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| EcoEA | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 5 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8582806 | large |
| EcoEA | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 2 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8603487 | large |
| EcoEA | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 289 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.6624961 | large |
| Fitness Sharing | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 768 | 9.02e-04 | 0.0225500 |
|
p = 0.02255 | 0.3322821 | moderate |
| Fitness Sharing | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 762 | 7.77e-04 | 0.0194250 |
|
p = 0.019425 | 0.3364184 | moderate |
| Fitness Sharing | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 809 | 2.39e-03 | 0.0597500 | ns | p = 0.05975 | 0.3040175 | moderate |
| Fitness Sharing | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 988 | 7.14e-02 | 1.0000000 | ns | p = 1 | 0.1806181 | small |
| Fitness Sharing | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1312 | 6.72e-01 | 1.0000000 | ns | p = 1 | 0.0427417 | small |
| Lexicase | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Lexicase | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 23 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8458717 | large |
| Random | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 0 | 0.00e+00 | 0.0000000 | **** | p < 1e-04 | 0.8617275 | large |
| Random | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1208 | 7.75e-01 | 1.0000000 | ns | p = 1 | 0.0289540 | small |
| Random | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1244 | 9.70e-01 | 1.0000000 | ns | p = 1 | 0.0041363 | small |
| Random | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1257 | 9.64e-01 | 1.0000000 | ns | p = 1 | 0.0048257 | small |
| Tournament | 10 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 394 | 0.00e+00 | 0.0000001 | **** | p < 1e-04 | 0.5901110 | large |
| Tournament | 100 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 482 | 1.00e-07 | 0.0000030 | **** | p < 1e-04 | 0.5294454 | large |
| Tournament | 1000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1107 | 3.26e-01 | 1.0000000 | ns | p = 1 | 0.0985816 | small |
| Tournament | 10000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1739 | 7.58e-04 | 0.0189500 |
|
p = 0.01895 | 0.3371078 | moderate |
| Tournament | 100000 | value | Phenotypic -> Phylogenetic | Phylogenetic -> Phenotypic | 50 | 50 | 1675 | 3.43e-03 | 0.0857500 | ns | p = 0.08575 | 0.2929873 | small |
References
Allen, Micah, Davide Poggiali, Kirstie Whitaker, Tom Rhys Marshall, Jordy van Langen, and Rogier A. Kievit. 2021. “Raincloud Plots: A Multi-Platform Tool for Robust Data Visualization.” Wellcome Open Research 4 (January): 63. https://doi.org/10/ghvbc6.
Hernandez, Jose Guadalupe, Alexander Lalejini, and Charles Ofria. 2021. “An Exploration of Exploration: Measuring the Ability of Lexicase Selection to Find Obscure Pathways to Optimality.” arXiv:2107.09760 [Cs], July. http://arxiv.org/abs/2107.09760.